Имуномедиирани възпалителни невропатии
-
Код:KS4183
Клинико-имунологични електрофизиологични корелации

Съдържание:
Използвани съкращения.6
Рецензии 8
Въведение 15
I ЧАСТ – Диагностика на полиневропатия 18
1. Анатомия на периферните нерви 18
2. Физиология на нервното провеждане 21
3. Клиничен подход при диагностика на полиневропатията 27
3.1. Времеви профил и прогресия на полиневропатията 28
3.2. Тип и размер на засегнатите нервни влакна 28
3.3. Патерн на засягане на нервните влакна 31
3.4. Подлежаща патология (аксонална, демиелинизираща или смесена) 32
3.5. Фамилна анамнеза за полиневропатия, минали заболявания, професионална увреда или интоксикация 33
4. Електрофизиологичен подход. 36
4.1. Фокусирана анамнеза и неврологичен статус 37
4.2. Планиране на електродиагностичното изследване 37
4.3. Неврография 38
4.3.1. Неврографски показатели на интактните нерви
40 4.3.2. Aксонална дегенерация 42
4.3.2.1. Критерии за аксонална полиневропатия 42
4.3.3. Демиелинизация .43
4.3.3.1. Демиелинизиращо забавяне на нервното провеждане 43
4.3.3.2. Проводен блок 45
4.4. Неврофизиологични критерии за ХВДП 47
4.5. Разпределение на промените при демиелинизиращи невропатии 48
5. Лабораторен диагностичен подход 51
5.1. Биопсия на нерв и кожа 51
5.1.1. Светлинно-микроскопско изследване на разслоени нервни влакна 53
5.1.1.1. Валерова дегенерация 53
5.1.1.2. Първична демиелинизация 54
5.1.2. Електронно-микроскопско изследване 56
5.2. Ликворно изследване 56
5.3. Изследване за моноклонална гамапатия с неопределена значимост 57
5.4. Гликопротеинови, ганглиозидни и сулфатидни антитела 57
5.4.1. Гликолипидни антитела при синдром на Guillain-Barré 59
5.4.2. Гликолипидни антитела при ХВДП 60
5.4.3. Диагностична стойност на антигликолипидните антитела 61
5.5. Ревматологични тестове 62
5.6. IgG4 антитела 63
4 ИМУНОМЕДИИРАНИ ВЪЗПАЛИТЕЛНИ НЕВРОПАТИИ II ЧАСТ – Guillain–Barré синдром 75
1. Варианти на Guillain–Barré синдром 75
1.1. Остра възпалителна демиелинизираща полиневропатия 77
1.1.1. Патохистологични и имунологични характеристики 77
1.2. Аксонални варианти на СГБ 80
1.2.1. Остра моторна и моторно-сетивна аксонална невропатия 80
1.2.2. Фаринго-цервико-брахиален вариант на СГБ 83
1.2.2.1. Клинична характеристика на болните с ФЦБ 85
1.2.2.2. Патофизиология на ФЦБ 85
1.2.2.3. Диагностични процедури при ФЦБ 87
1.2.3. Miller Fisher синдром 88
1.2.4. Остри атаксични невропатии, асоциирани с дизиалозилови антитела 92
2.0 Клинико-електрофизиологични корелации при СГБ 95
2.1. ОВДП и демиелинизиращ блок в провеждането 95
2.2. ОМАН и аксонална дегенерацияю 98
2.3. ОМАН с обратим блок в провеждането 101
2.4. Елекрофизиологични критерии за СГБ 101
3. Диференциална диагноза 102
3.1. Атипичен СГБ 103
3.2. Диференциална диагноза 103
3.3. СГБ, асоцииран с COVID-19 104
3.4. Потенциална роля на ваксинацията за СГБ 107
III ЧАСТ – Хронична възпалителна демиелинизираща невропатия 121
1. Патохистологични характеристики на ХВДП 122
2. Имунопатогенеза на ХВДП 122
2.1. Клетъчен имунен механизъм при ХВДП 122
2.1.1. Роля на кръвно-нервната бариера 123
2.1.2. Възпалителни инфилтрати 124
2.1.3. Роля на CD8+ T клетки 124
2.1.4. Т-регулаторни клетки и централен имунен толеранс 125
2.1.5. Роля на макрофагите в имунопатогезата на ХВДП 126
3. Клинични характеристики на ХВДП 129
3.1. Типичен ХВДП 131
3.1.1. Tипичен ХВДП: преференциално засягане на дисталните терминали и проксимални нервни участъци 131
3.1.2. Tипичен ХВДП: преференциално засягане на моторни нерви 133
3.2. ХВДП варианти 134
4. Диференциална диагноза 140
4.1. Имитиращи ХВДП невропатии 140
4.1.1. Парапротеинемична невропатия 140
4.1.1.1. IgM парапротеинемична дистална невропатия 143
4.1.1.2. CANOMAD невропатия 148
Съдържание 5 4.1.1.3.
POEMS невропатия 150
4.1.2. Мултифокална моторна невропатия 152
4.1.2.1. MMН: електродиагностични промени 155
4.1.2.2. Други диагностични методи за ПБ при MMН 161
4.1.3. Демиелинизираща Charcot-Marie-Tooth невропатия 162
4.1.4. Амилоидна невропатия 163
4.2. Асоцииран с други заболявания ХВДП 163
4.2.1. Диабет и ХВДП 164
4.2.2. Charcot-Marie-Tooth и ХВДП 165
4.2.3. Други автоимунни заболявания и ХВДП 165
4.2.4. Злокачествени и инфекциозни заболявания и ХВДП 166
4.2.5. Медикаменти и ХВДП 166
5. Терапевтично поведение при ХВДП 167
IV ЧАСТ – Концепцията нодо-паранодопатия 190
1. Нодопатии 190
1.1. Антиганглиозидни антитела 190
1.2. Антитела срещу нодални протеини 192
1.3. Нодопатии: обратим аксонален блок в провеждането 192
2. Паранодопатии 194
2.1. Анти-N155 антитела 194
2.2. Анти-CNTN1 антитела 195
2.3. Анти-CASPr антитела 196
3. Концепцията „нодо-паранодопатия“ 197
4. Роля на субкласовете IgG имуноглобулини 197
V ЧАСТ – Вместо епилог: електродиагностични проблеми, нови диагностични и терапевтични стратегии 203
1. Електродиагностични проблеми при Guillain–Barré синдром 203
2. Електродиагностични проблеми при ХВДП 206
3. Нови диагностични техники 207
3.1. Напреднали електродиагностични техники 207
3.1.1. Оценка на аксонната възбудимост 207
3.1.2. Оценка на моторната скорост на провеждане чрез SF-EMG електроди 212
3.2. Магнитно-резонансна неврография и ултразвуково изследване на периферни нерви 215
3.3. Напреднали биопсични техники 219
3.3.1. Таргетна фасцикуларна биопсия 219
3.3.2. Биопсия от кожа: оценка на промените на нодалната архитектоника 222
4. Нови терапевтични стратегии 223
4.1. Терапевтично приложение на инхибитори на комплемента 223
4.2. Подобрение на терапевтичната ефективност на IVIg 224
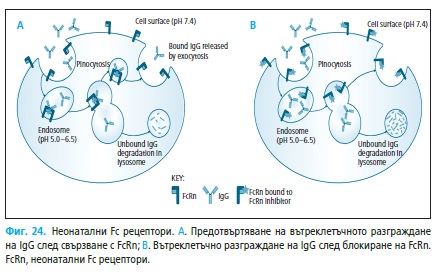
4.3. Протеозомни и FcRn инхибитори
Въведение:
Така се случи, че последните десет години от професионалната ми кариера преминаха като консултант по клинична неврофизиология във Великобритания, но положих неимоверни усилия да не прекъсвам клиничната си практика в България. Разделен между родината и чужбина, имах (и имам) редкия шанс да се сблъсквам с диагностичните предизвикателства на болните с периферно-нервни заболявания, консултирани в университетските центрове на Лондон, Единбург, Бирмингам, Ковънтри, Лестър и Челмсфорд, амбулаторните практики във Видин и Плевен. В първите години като консултант във Великобритания бях изненадан от необичайно големия брой на болните с хронична възпалителна демиелинизираща полиневропатия (ХВДП). Първоначално предполагах, че причината се крие в клъстера на тези пациенти, насочени за електродиагностично изследване в третичните центрове. По-късно осъзнах, че има и друго обяснение. Няколко години по-рано стартира програма за лечение на ХВДП с интравенозен имуноглобулин. На повторна електродиагностична оценка подлежаха случаите, показали недостатъчен ефект от лечението. При някои се намираше тежка аксонална загуба, а други (не малка част) не покриваха електродиагностичните критерии за демиелинизация. При прегледите у нас се сблъсквах с противоположен проблем. Диагностицирани като години за хронични радикулопатии (по-рядко тунелни невропатии) болни, се оказваха типични случаи на ХВДП. За второ мнение към мен се обръщаха и близки на болни със синдром на Guillain– Barré, неповлияни от прилаганата доза имуновенин, което ме изправяше отново пред предизвикателствата на родната действителност и ежедневните проблеми на моите колеги. И така с течение на годините ясно се очертаха два противоположни, но с еднаква важност диагностични проблема – хипердиагностика на ХВДП в чужбина и суб(не)диагностициране на това заболяване у нас. Всеки един от тях има своето обяснение.
Демиелинизиращите невропатии съставляват малка част (около 15%) от невропатиите, но техните възпалителни форми имат съществено предимство. Това са потенциално 16 ИМУНОМЕДИИРАНИ ВЪЗПАЛИТЕЛНИ НЕВРОПАТИИ лечими състояния и тяхната хипердиагностика може да бъде оправдана с естествения вътрешен стремеж на лечителя да лекува (въпреки отбелязания в последното десетилетие напредък неврологичната специалност е длъжник на пациентите в това отношение). Анализът на литературата ме убеди, че хипердиагностиката на ХВДП е световен проблем (18, 36). Обективна причина за това е отсъствието на „златен“ диагностичен стандарт. Разработени са повече от 15 групи диагностични критерии (подробно описани в настоящата монография), всеки един със своите предимства и недостатъци. Основно място в диагностиката на невропатиите заема електродиагностичното изследване. Техническите затруднения, съпътстващи това изследване, както и неправилното интерпретиране на резултатите обуславят значителна част от диагностичните грешки. Всичко това ме мотивира да предприема настоящия труд. Заглавието на монографията включва термина „имуномедиирани възпалителни невропатии“, като под такива разбирам невропатиите, при които възпалителният процес се тригерира и поддържа от имунни механизми (автоантитела, имуномимикрия, комплемент-медиирана увреда и т.н.) и води до дегенеративни промени в нервните влакна (демиелинизация или аксонална дегенерция). Изкушавах се вместо „имуномедиирани“ да използвам „автоимунни“, но не при всички форми са идентифицирани специфични антитела. Напредъкът в експерименталните проучвания ми дава надежда, че в близко бъдеще използването на подобна терминология ще е напълно оправдано.
В първата част на монографията е представен утвърденият от водещи специалисти алгоритъм при диагностика на невропатия. Описани са специфичните патерни, отразяващи локализацията и обхвата на засягане на периферните нерви, времевия профил, генетичната и професионална предразположеност. Особено място е отделено на интерпретацията на данните от електродиагностичното изследване, в детайли са представени промените, настъпващи при демиелинизация и аксонална дегенерация, както и общоприетите критерии за тяхното разграничаване. Съгласно установените у нас традиции острите и хронични форми на възпалителни невропатии се представени самостоятелно в отделни части на монографията, подробно са описани техните клинични, електродиагностични и имунологични варианти. Представени са характеристиките на парапротеинемичните невропатии, както и някои специфични парапротеинемични синдроми – POEMS и CANOMAD, диагностичните Въведение 17 затруднения и решения при разграничаване на ХВДП от неговите варианти. Специална част от монографията е отделена на нодо-паранодопатиите. Тези наскоро описани възпалителни невропатии, чиято класификация се основава по-скоро на локализацията, отколкото на специфичната подлежаща патология, показват клинични, електродиагностични и най-вече терапевтични особености, отличаващи ги от останалите възпалителни форми. Неоспорим факт е, че практикуващият клиницист рядко посяга към белия лист. Това има своите оправдания. Времето му е постоянно заето от консултации, дежурства, пътувания.
Трудностите, наложени от ковид пандемията, ми осигуриха необходимото време за обобщение на отдавна загнездени в съзнанието ми клинични проблеми, касаещи имуномедиираните възпалителни невропатии. В настоящата монография са представени първите публикувани случаи на увреда на периферните нерви, „причинени“ от или „съпровождащи“ COVID-19 инфекцията. Има и друга причина за положените от мен усилия. Правилата на добрата практика в чужбина налагат клиницистите непрекъснато да се учат от собствените грешки (принцип, дълбоко залегнал в народопсихологията, но не намерил особено място в клиничната практика у нас). Бих искал да уверя младия читател, разгръщащ страниците на този скромен труд, че голяма част от заключенията (обобщени в края на всяка част) са резултат от предизвикателствата, пред които се изправях, и грешките, които допусках. Налагаше се да се коригирам, понякога разчитайки на прочетеното, наученото, неврологичното чукче и електромиографския апарат, а друг път на мъдростта и споделения опит от колеги и приятели. Клиничната практика може да е успешна, единствено когато следва постоянно променящите се нужди на пациентите, отразени в усвояването и внедряване на нови диагностични и терапевтични умения. С част от тях читателят може да се запознае в последната част на монографията. Скромният опит на клиницист, изследовател и преподавател ме е убедил, че прозрение на клиничните проблеми най-добре се постига по време на обучението на нашите ученици. Уверявам читателите, че и аз самият много научих, докато се трудих над настоящата монография, и им пожелавам приятно четене.
От автора 12.01.2021 г. Челмсфорд, Великобритания
I ЧАСТ
Диагностика на полиневропатия
Клиничното изследване (анамнеза и неврологичен статус) при болни
с полиневропатия и в частност имуномедиирана невропатия осигурява
информация за общите белези и симптоми, но не дава отговор за подле-
жащата патофизиология и не e особено полезно при определяне давност-
та и разпределение на периферно-нервните лезии.
Електродиагностичното изследване позволява по-дейтално характе-
ризиране и класифициране на полиневропатиите. Надеждна интерпрета-
ция на резултатите е възможна единствено в контекста на клиничната на-
ходка и при задължително отчитане влиянието на техническите фактори.
Диагностичният подход при невропатиите е комплексен и изисква
анализ на клинични, електрофизиологични, патохистологични и имуно-
логични данни. Най-важни са белезите от клиничното и електрофизио-
логично изследване, сравнително достъпна диагностична процедура с
добра чувствителност и специфичност при полиневропатия. При недос-
татъчни или противоречащи си клинични и електрофизиологични резул-
тати, диагнозата се установява с помощта на нервна биопсия или чрез
специфични лабораторни изследвания.
Интерпретацията на клиничните и електродиагностични резултати
е възможна, само ако детайлно се познават анатомичните и физиологич-
ни характеристики на периферните нерви, специфичните имунни меха-
низми и патологични промени в периферните нерви в резултат от имун-
ната атака.
1. Анатомия на периферните нерви
Индивидуалните периферни нерви се отделят от плексусите, като по-
вечето от тях са смесени и се състоят от сензорни, моторни и автономни
нервни влакна.
Кожните сензорни нерви са отделни нервни клончета, които се присъ-
единяват към смесените нерви обикновено от дисталните кожни области,
а някои от тях от по-проксимални места (напр. кожни сензорни клончета
в предмишницата). Всеки сензорен нерв съдържа от 3000 до 6000 нервни
влакна.
Моторните нерви инервират групи от мускули, каквито са например
тенарната и хипотенерна група. Те пенетрират в мускула през една или
няколко моторни точки, след което се разклоняват на множество дистал-
ни клончета, завършващи в 500 до 800 дистални терминали. Дисталните
терминали са немиелинизирани, а дисталните нервни клончета (подоб-
но на нервните коренчета) се характеризират с кръвно-нервна бариера,
не толкова ефективна, колкото тази в ствола на периферния нерв (106).
„Моторните“ клончета в мускулите съдържат моторни и аферентни Ia
сетивни влакна (започващи от мускулните вретена и сухожилия) в съ-
отношение приблизително 50% на 50%, поради което наименованието
„моторно“ клонче не е точно от анатомична гледна точка. При лезии на
моторните нерви дефицитът изглежда чисто двигателен, тъй като кли-
нична оценка на функцията на аферентните Ia сетивни влакна е невъз-
можна (28).
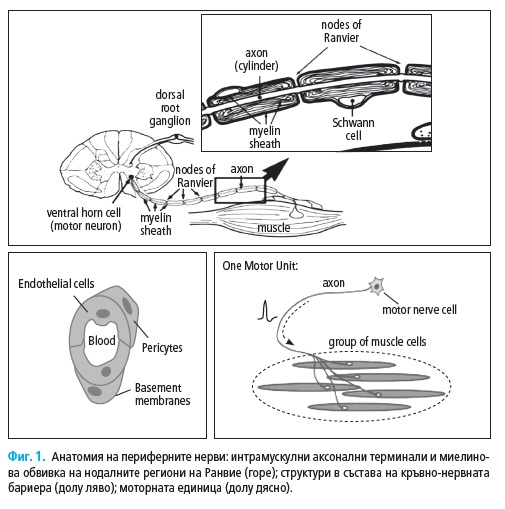
Моторната единица се състои от тялото на неврона, неговия аксон,
терминалните нервни клончета и инервираните от тях мускулни влакна
(Фиг. 1). Терминалните нервни клончета и мускулните влакна формират
в мускулите сферични области с диаметър от 5 до 10 mm. В мускулите с
повече от 100 моторни единици, териториите на 20 от моторните едини-
ци се припокриват в дадено място на мускула (28, 104).
Всяко нервно влакна се състои от аксон и миелинова обвивка, която
може да е дебела и плътна при влакната с голям диаметър или тънка и
не толкова плътна при тези с малък диаметър. В резултат от вариации-
те в диаметъра и степента на миелинизация нервните влакна провеж-
дат нервни импулси с различна скорост. Миелинизираните влакна имат
високи скорости на провеждане поради „скокообразното“ (салтаторно)
провеждане (30 – 60 m/s), докато немиелинизираните влакна провеждат
много бавно (< 1 m/s).
Рутинните неврографски изследвания оценяват основно дебелите
миелинови влакна, тъй като приносът на тънките миелинизирани и не-
миелинизирани влакна за формиране на сумарния отговор е нищожен.
Специфични неврографски тестове позволяват да се оцени интегрите-
та на тънките влакна, но тези процедури не се използват в ежедневната
практика и на практика нямат диагностична стойност при полиневропа-
тите (28, 104).
Периферните нерви с голям диаметър преди разклоняване в крайните
органи съдържат различни по размер и характер нервни влакна (дебели,
тънки, соматични, автономни, сензорни и моторни), след което моторни-
те и сензорни нерви влакна се разделят и стават достъпни за неврограф-
ско изследване (1, 43).
2. Физиология на нервното провеждане
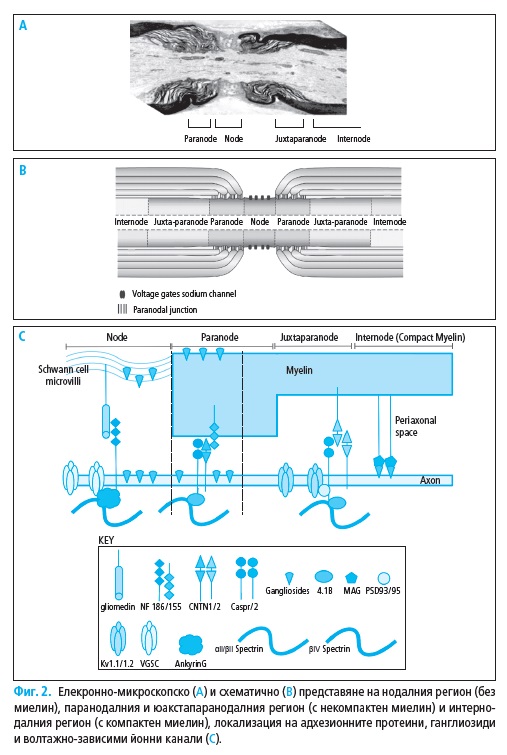
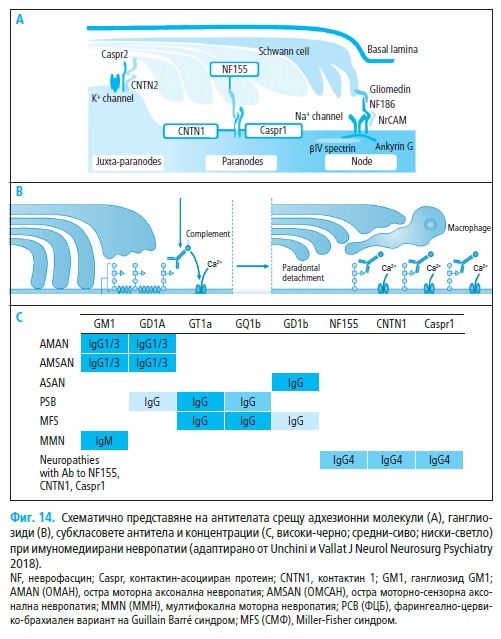
Миелинизираните нервни влакна са организирани в специфични ре-
гиони: нодус, паранодус, юкстапаранодус и интернодус (Фиг. 2). В но-
далните участъци (с дължина около 1 μm) миелинът е прекъснат и аксо-
лемата влиза в директен контакт с екстрацелуларното пространство, но
все още остава покрита от микровилите на Швановите клетки. В съсед-
ния паранодален участък, некомпактните миелинови ламели са плътно
прикрепени към аксолемата, а в областта на интернодуса (с дължина 1
– 2 mm) аксонът е заобиколен от компактен миелин.
Аксолемата на миелинизираните аксони при бозайниците има висо-
ко организирани молекулярни структури с нееднакво разпределение на
йонните канали (Фиг. 2).
Нодалните участъци показват висока плътност на волтажно-зависи-
ми (т.е. регулирани) Na+ каналчета (1000–2000/μm2) и бавни K+ каналче-
та. Юкстапаранодалният регион имат висока плътност на волтажно-за-
висими бързи K+ каналчета. Интернодалните участъци имат най-голям
абсолютен брой Na+ каналчета, бързи и бавни K+ каналчета, но тяхната
гъстота < 25/μm2 не е достатъчна за подържане пропагацията на акцион-
ния потенциал. Na+ каналчета са от Nav1.6 тип и осъществяват протича-
нето на транзиторен или персистиращ йонен ток. Няколко субтипа Na+
каналчета са представени в периферната нервна система, но нодусът на
възрастните бозайници съдържа единствено такива, които са Nav1.6 тип
(32).
Мембранният потенциал в покой в човешките аксони е −75 до −80
mV. Той зависи най-вече от равновесния потенциал на K+ йони, изтича-
щи навън през протеиновите пропускащи каналчета, и в по-малка степен
от равновесния потенциал на Na+ йони и Na+/K+ помпа (133).
Терминът деполяризация означава промяна на мембранния потен-
циал към по-позитивни стойностите от тези на мембрания потенциал
в покой за разлика от хиперполяризация, показваща преминаване към
по-негативни нива.
Транзиторните Na+ каналчета генерират 98–99% от общия нодален
йонен ток в посока към вътрешността на клетката, водещ до деполяриза-
ция на мембраната и генериране на акционен потенциал.
Персистиращите Na+ канали остават отворени по време на широките
колебания на мембранния потенциал и модифицират прага за възникване
на акционен потенциал (133).
Бързите K+ каналчета на юкстапаранодуса се отварят твърде скоро
(0,5 – 1,0 ms) след нодалните транзиторни Na+ каналчета. В резултат от
K+ йонен ток навън следва възвръщане на стойностите на мембранния
потенциал до тези на покой или дори хиперполяризация на мембраната.
В състояние на хиперполяризация мембраната е практически невъзбу-
дима и настъпва блокиране на последващите нервни импулси. Поради
тази причина в здравите нерви локализацията на бързите K+ каналчета е
отдалечена от нодуса – в юкстапаранодуса. Тези каналчета са анатомич-
но и функционално сепарирани от нодуса чрез комплекс от адхезионни
протеини, представляващи вертикална връзка между миелина и аксоле-
мата (Фиг. 2).
Бавните K+ каналчета са разположени в нодалните региони и се отва-
рят бавно (десетки милисекунди след настъпване на деполяризация). По
този начин се предотвъртява генериране на повторни акционни потен-
циали и индуциране на реполяризация в отговор на пролонгирана депо-
ляризация.
Na+/K+ помпа възстановява трансмембранния градиент на Na+ и K +
йони и подържа негативните нива на мембранния потенциал, като при
всеки един неин цикъл от аксона се отделят 3 Na+ и се вкарват 2 K+ йони
(133). Помпата се нуждае от енергия, при всеки цикъл се хидролизира по
1 молекула ATP. Локализацията на Na+/K+ помпа остава спорна.
Na+/Ca2+ помпа изхвърля Ca2+ от аксоплазмата, като ги разменя за Na+
и по този начин подържа Ca2+ хомеостаза (133). Когато вътреклетъчната
концентрацията на Na+ достигне критични нива, Na+/Ca2+ помпа обръща
посоката си на действие, което води до увеличение на интрацелуларните
Ca2+ нива (133).
В нодалния участък Na+ волтажно-зависими каналчета са свързани с
анкирин G, който от своя страна се прикрепя към адхезионните проте-
ини спектрин и неврофасцин-186. Последният е свързан с молекулата на
глиомедин, локализирана върху микровилите на Швановите клетки (112).
В паранодуса аксонните адхезионни протеини – контактин-свър-
зан протеин-2 и контактин-1 са плътно прикрепени към миелиновия
адхезионен протеин неврофасцин-155. По този начин се формира ком-
плексна и здрава аксонно-глиална вертикална връзка, сепарираща клъс-
терите от Na+ каналчета на нодуса от бързите K+ волтажно-зависими ка-
налчета на юкстапаранодуса.
В юкстапаронодуса K+ каналчета са прикрепени към аксонните
адхезионни протеини – контактин-свързан протеин-2 и котактин-2.
Контактин-свързаният протеин-2 (известен още като транзиторен аксона-
лен гликопротеин-1) е експресиран както върху аксолемата, така и върху
противостоящата Швановата мембрана. Взаимоотношенията между кон-
тактин-свързания протеин-2 и аксонно-миелиновия контактин-2 играят
важна роля за клъстера на волтажно-зависимите K+ каналчета (Фиг. 2).
Ганглиозидите са гликолипиди, състоящи се от липиден компонент
церамид (локализиран в мембраната) и екстрацелуларна въглехидратна
група, към която се прикрепят 0, 1 или повече групи сиалова киселина.
Светлинно-микроскопското изследване с висока резолюция на спинални
коренчета и седалищен нерв на плъхове показва, че GM1 ганглиозидите
са локализирани върху нодалната и паранодална аксолема, абаксонална-
та (външна) мембрана и микровилите на Швановите клетки. GD1a ганг-
лиозиди са локализирани в нодалните аксонални региони и абаксонална-
та Шванова мембрана (51). Ганглиозидите взаимодействат с нодалните
протеини и подържат аксонно-глиалния стабилитет на паранодуса (Фиг.
2). В експериментални модели на генно-променени мишки GM1 и GD1a
ганглиозиди не успяват да се свържат аксолемата, което корелира с тежко
нарушение на клъстера от Na+ каналчета и изместване на K+ каналчета
към паранодуса (120).
В активирания нод при достигане на необходимия праг на деполяри-
зация, волтажно-зависимите Na+ каналчета се отварят, настъпва инфлукс
на Na+ йони (акционен ток) и възниква акционен потенциал (потенциал
на действие; Фиг. 3). Повишеният позитивен товар от вътрешната стра-
на на вече деполяризираната мембрана води до поява на електроничен
потенциал, предизвикващ пасивно придвижване на позитивните товари
към вътрешната страна на подлежащия на деполяризация нодус (раз-
пространяващ се потенциал; Фиг. 4). В същото време отрицателно на-
товарената външна страна на вече деполяризираната мембрана привлича
позитивни товари от външната страна на подлежащия на деполяризация
нодус. Неговите Na+ каналчета са затворени и акционен ток е невъзмо-
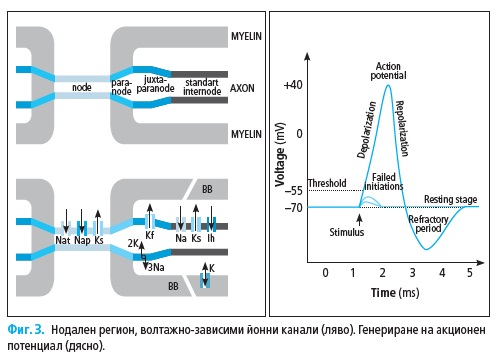
жен. Ако разпространяващият се потенциал (постепенно намаляващ по
хода на аксона) успее да достигне деполяризационния праг, волтажно-за-
висимите Na+ каналчета се отварят и възниква насочен към вътрешността
на аксона акционен ток. Последвият играе ролята на своеобразен бустер
за постепенно намаляващия разпространяващ се потенциал (46).
Факторът сигурност (ФС) е алгебрична дроб, чиито числител – по-
тенциалът на разпространяващия се ток е разделен на деполяризацион-
ния праг на подлежащия на деполяризация нодус (122). В интактните
нерви стойностите на ФС са между 5 и 7. Това означава, че за да се гаран-
тира безпроблемно провеждане е необходимо генериране и подържане
на мощен разпространяващ се ток, който 5 до 7 пъти да надвишава прага
на деполиризация на подлежащия на активация нодус. При увреждане
на нервите ФС намалява, а при стойности на ФС < 1 провеждането на
нервния импулс е блокирано.
Промените на ФС могат да се дължат на различни патологични про-
цеси: дисфункция или деструкция на нодалните Na+ каналчета, интерно-
дална демиелинизация, паранодална и юкстапаранодална „демиелиниза-
ция“ (отлепване на миелина).
При имуномедиирана атака на нодуса, предизвикана от антигангли-
озидни или насочени срещу адхезионните протеини антитела, броят на
Na+ каналчета временно (дисфункция) или перманентно (деструкция)
намалява, което затруднява генерирането на акционния потенциал. При
интернодална демиелинизация разпространяващият се от вътрешната
страна на аксоналната мембрана потенциал към подлежащия на деполя-
ризация нодус се изчерпва по хода на аксона поради изтичане на пози-
тивни товари през порите на миелиновите лезии, което води до редукция
на ФС (46, 47). При отлепване на паранодалния миелин подлежащият на
активация регион е патологично разширен и включва както нодалния,
така и съседния паранодус. Поради отлепване на миелина („демиелини-
зация“) тази зона е не само разширена, но и с повишена капацитивност
(повишена сила на привличане на противоположно натоварени йони то-
вари от двете страни на мембраната), затрудняваща или блокираща про-
тичането на разпространяващия се електроничен ток от външната стра-
на на аксоналната мембрана към вече деполяризирания нодус. Ако този
участък все пак успее да се деполяризира, това е за сметка на повече от
нормалното за това време и се съпровожда от значително забавяне на
нервното провеждане (46). При демиелинизация на юкстапаранодусна
настъпва бързо блокиране на нервната проводимост, тъй като в резул-
тат от активиране на бързите K+ каналчета на юкстапаранодуса настъп-
ва преждевременна реполяризация, хиперполяризация и инактивация
на Na+ нодални каналчета. Провеждането е допълнително смутено и от
много високата капацитивност на извънредно разширената и лишена от
миелин юкста-паранодо-нодална зона (46).
Въпреки различните механизми, обуславящи промените на ФС при
различни патологични локализации, неврографското изследване, което
отразява функционалното състояние на всичките стимулирани аксони и
интернодални сегменти показва сравнително ограничен брой промени –
ниска моторната скорост, увеличена дисперсия на моторните отговори и
проводен блок (47).
II ЧАСТ
Остри възпалителни
имуномедиирани невропатии
Синдромът на Guillain–Barré (СГБ) e остро имуномедиирано забо-
ляване на периферните нерви, което все още е най-честа причина за по-
стинфекциозна невромускулна парализа по целия свят. Неговата заболе-
ваемост между 0,6 и 4 случая на 100 000 население (67). Вероятността за
отделният индивид в един момент от живота си да се разболее от GBS е
средно 1:1000 (162).
Терминът СГБ дълго време се считаше за синоним на остра възпа-
лителна демиелинизираща полирадикулоневропатия (ОВДП), но на-
последък епонимът СГБ се използва за обозначаване и на аксоналните
субтипове, които имат различен патофизиологичен и имунологичен ме-
ханизъм.
1. Guillain–Barré синдром
В 1859 г. Landry за първи път описва невропатия, характеризираща се
с остра асцендираща парализа. По-късно Guillain, Barré и Strohl добавят
асоцииращите се с невропатията арефлексия и албумино-клетъчна дисо-
циация в ликвора (55). Участието на Landry и Strohl e незаслужено игно-
рирано и по-късно синдромът придобива известност само като синдром
на Guillain–Barré.
В 1949 г. Haymaker и Kernohan описват хистопатологичните белези
на 50 случаи на СГБ с фатален край (61). Ранните (в рамките на първата
седмица) патологични промени включват оток на проксималните нерви
и последваща дегенерация на миелиновите обвивки. В първите съобще-
ния обаче не са описани възпалителни инфилтрати. Такива са устано-
вени по-късно. Наличието на манифестни периваскуларни васкуларни
инфилтрати в спиналните коренчета, дорзалните ганглии, краниалните
нерви, по-рядко по хода на периферните нерви и едновременно с това
разположени в непосредствена близост с участъци на сегментна демие-
линизация (9) дават основание полиневропатията да бъде наречена остра
възпалителна демиелинизираща полирадикулоневропатия (ОВДП) (16,
78, 130, 131).
В последните десетилетия се установи, че СГБ не е хомогенно забо-
ляване а по-скоро синдром, състоящ се от няколко субтипа остри имуно-
медиирани полиневропатии (6, 131). В допълнение към ОВДП се доба-
виха двете аксонални форми на СГБ – остра моторно-сензорна аксонална
невропатия (ОМСАН) и остра моторна аксонална невропатия (ОМАН).
По-късно за още две заболявания, чиято клинична картина е различна от
тази на ОВДП, синдрома на Miller Fisher (СMФ) и острата сензорна аксо-
нална невропатия се установи, че имат сходна имунопатогенеза, поради
което могат да се разглеждат като вариант на аксоналния СГБ.
1.1. Остра възпалителна демиелинизираща полиневропатия
1.1.1. Патохистологични и имунологични характеристики
Нервната биопсия не е рутинна диагностична процедура при СГБ,
поради което патохистологичните познания за това заболяване се бази-
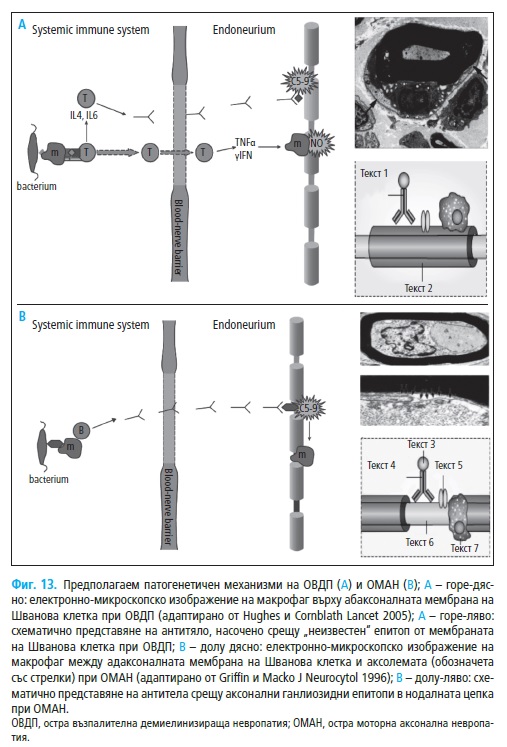
рат основно на находките при аутопсионни изследвания (Фиг. 13A).
Първите патохистологични описания датират от 1969 г., когато
Asbury и съавт. описват описват клиничните и патоанатомични промени
при 19 случаи на СГБ с фатален край (9). Болшинството от починалите са
имали бързо прогресиращ неврологичен дефицит, който в рамките на 3
до 7 дни се усложнява с респираторна парализа. Обща патохистологична
находка е наличие на възпалителен демиелинизиращ неврит и развитие
на фокална периваскуларна лимфоцитна инфилтрация, засягаща пери-
ферната нервна система на всички нива. Възпалителните инфилтрати са
били добре оформени при всички случаи, включително и при починали
в рамките на няколко дни. Сегментна демиелинизация е описана в зони,
които кореспондират с възпалителните инфилтрати. Описана е различ-
на по степен Валерова дегенерация, варираща в съответствие с тежестта
на демиелинизиращите лезии. Клиничните симптоми корелират с лока-
лизацията на патологичните промени. Периваскуларните възпалителни
инфилтрати персистират в продължение на месеци и дори години след
отзвучаване на клиничната симптоматика.
Последващите светлиномикроскопи изследвания при СГБ потвържда-
ват разположените периваскуларно клетъчни инфилтрати, съставени ос-
новно от макрофаги и лимфоцити (65, 76, 100, 131). Те са разположени
предимно в нервните коренчета и в предразположени към тунелни нев-
ропатии нервни участъци, както и в крайните интрамускулни разклоне-
ния на моторните нерви. Най-ранните хистологични промени настъпват
в прищъпванията на Ranvier. Първоначално се наблюдава отлепване на
паранодалния миелин, след което в интернодалните региони се развива
класическа демиелинизация. В участъците на сегментна демиелиниза-
ция се установява мононуклеарна инфилтрация. Заедно с моноцитите се
откриват полиморфонуклеарни левкоцити, които в тежките случаи пред-
хождат развитието на аксонална дегенерация. По време на възстанови-
телната фаза се наблюдават белези на ремиелинизация, които се харак-
теризират с поява на увеличен брой (в сравнение с незасегнатите нерви)
интернодалните сегменти с „изтънен“ миелин.
Промените при СГБ твърде много наподобяват тези при екперимен-
тален алергичен неврит (EAН), модел за отсрочена хиперсензитивност,
медииран от специфично активирани лимфоцити. При EAН активирани-
ят T клетъчно-медииран имунен отговор играе основна патогенетична
роля. При СГБ (подобно на EAН) сe позитивират серумните маркери за Т
клетъчна активация (разтворим интерлевкин-2 рецептор и интерферон)
(131, 162). Неизменна находка при EAН е присъствието на макрофаге-
ални клетки, инвазиращи обвивките на аксоните. Те са насочени срещу
повърхностни антигени на Швановите клетки и миелина, което от своя
страна води до последваща активация на T клетките (49).
На основа на патохистологичните прилики с ЕAН Asbury и съавт.
(1969) правят извода (9), че ОВДП-СГБ е клетъчно-медиирано заболя-
ване, при което перифернонервната тъкан и по-специално миелина са
атакувани от активирани специфични Т лимфоцити. Този извод, напра-
вен преди повече от 50 години, е възприет от повечето изследователи и
доскоро не беше подлаган на съмнения. В последните години обаче се
установи, че хуморалният имунен отговор е също ангажиран в патоге-
незата на ОВДП. Това твърдение се подкрепя от увеличените серумни
нива на различни антиганглиозидни антитела, а също така и от наблю-
даваното клинично подобрение след плазмафереза (59, 60). Инжектира-
нето на серум от пациенти с ОВДП в нерви на експериментални модели
индуцира комплемент-медиирана демиелинизация и проводен блок (38).
Buchwald и съавт. (1998) изследват ефекта от инжектиране на серум от
пациенти със СГБ върху диафрагми на мишки и установява инхибиране
на пресинаптичното невромускулното предаване (19). В друго проучване
са намерени циркулиращи серумни IgG антитела срещу Шванови клетки
при ¼ от болните със СГБ (97). Чрез имунофлуоресцентна микроскопия
промените при СГБ са локализирани в дисталните части на микровилите
на Швановите клетки. Според Kwabara и съавт. (2004) имунната атака
е насочена срещу немиелинови (най-вероятно гликолипидни) епитопи и
протеини, свързващи Швановите клетки с аксолемата (93).
Положителният ефект от прилагането на плазмафереза (ПФ) при
СГБ показва, че хуморални фактори (антитела и комплeмент) играят ва-
жна роля в неговата патогенезата (в т.ч. и при ОВДП), тъй като терапев-
тичният ефект от ПФ се свързва с отстраняване на хуморални фактори
(а не на Т клетки). Секретираните от T клетките цитокини също биха
могли да бъдат отстранени при ПФ, но плазменият им полуживот е само
няколко часа и ако ефектът от ПФ се дължи на тяхното елиминирането,
би трябвало да е краткосрочен (а е добре известно, че не е).
Патохистологични проучвания в ранни стадии на ОВДП демонстри-
рат активация на комплемента по външната повърхност на Швановите
клетки, където се наблюдават и ранни везикуларни миелинови дегене-
рации. Имунохистохимични изследвания обаче не потвърждават специ-
фично отлагане на имуноглобулини, което най-вероятно се дължи на спе-
цификата на ендоневралното оцветяване (58).
Въпреки че хипотезата за активиране на клетъчния имунитет не е
напълно отхвърлена, описаните промени недвусмислено говорят в полза
на обратното, а именно че първичните промени при ОВДП се дължат
на свързване на автоантитела към неопределени (за сега) епитопи от
абаксоналната (външна) мембрана на Швановите клетки, което води до
последваща активация на комплемента. Предизвиканата по този начин
демиелинизация може да е дифузна по протежение на нервните стволо-
ве, но в ранната фаза е обикновено ограничена в нервните коренчета и
дисталните интрамускулни клончета на нервите (където кръвно-нервна-
та бариера не е толкова ефективна). Последва пролиферация на Швано-
ви клетки, които мигрират към местата на демиелинизация и ремиели-
низират уголените аксони, причина за клиничното възстановяване при
ОВДП. Аксоните също могат да бъдат засегнати от болестния процес,
но аксоналната увреда е вторична на демиелинизацията и обикновено се
съпровожда от тежка инвалидност (147).
1.2. Аксонални варианти на СГБ
1.2.1. Остра моторна и моторно-сетивна аксонална невропатия
За първи път в 1996 г. Hafer-Macko и съавт. описват натрупване на
IgG и активиране на комплемента в аксолемата на болни с остра мотор-
на аксонална невропатия (ОМАН). Депозирането на тези възпалителни
компоненти предхожда развитието на аксонална дегенерация (Фиг. 13В).
Засягат се нодалните региони, където към С5а фракцията и другите ком-
поненти на комплемента се прикрепят макрофагеални клетки. Последва
прекъсване на влакната в нодалните региони, след което макрофагеални-
те клетки се преместват към интернодалните периаксонални простран-
ства. Миелиновите обвивки на засегнатите влакна остават интактни, но
аксоните изглеждат изместени встрани от локализираните между Шва-
новите клетки и аксолемата макрофагеалните инфилтрати. При по-тежки
случаи настъпва увреждане на голям брой нервни влакна и като цяло
находките недвусмислено показват, че ОМАН е IgG комплемент-медии-
рано заболяване с таргетни епитопи, локализирани в аксолемата на мо-
торните влакна (58).
Депозирането на IgG и комплемента при ОМАН се различава от това
при ОВДП. Както се отбеляза при ОВДП маркерите за активация на ком-
племента C3d и С5b-9 са локализирани в близост с абаксоналната (външ-
но разположена) мембрана Швановите клетки (58). При ОМАН компле-
ментът обаче е активиран в непосредствена близост с адаксоналната
(вътрешна, периаксонална) мембрана, най-често в нодалния и интерно-
дален регион, като до този момент при ОМАН не са описани транзитор-
ни случаи по отношение на локализацията. Периаксонална лимфоцитна
инфилтрация и демиелинизация се установяват единствено в случаи с
изразена аксонална дегенерация (15). Таргетите на имунната атака са
локализирани в нодалните участъци на моторните влакна, най-вероятно
Gal(p1-3)GalNAc ганглиозидни епитопи. Към него се свързват анти-GM1
антитела (53), установени в серума на част пациентите с аксонални фор-
ми на СГБ (52, 165).
Случаите на ОМАН най-често последват инфекция от Campilobacter.
Мембраните на изолирани микроорганизми показват експресия на
Gal(p1-3)GalNAc, липозахаридни компоненти в състава на GM1 ганглио-
зидите и най-вероятно липополизахаридите индуцират имунния отговор
към GM1 (Фиг. 14). Анти-GM1реактивност е установена и при микроор-
ганизми, непоказващи липополизахаридни характеристики, поради кое-
то се предполагат и други механизми за продукция на анти-GM1 анти-
тела (53, 123), като напр. антигенни детерминанти в нодалните възли на
моторните влакна (45, 133, 142, 143).
Само няколко месеца след като Hafer-Macko и съавт. (1996) докладват
първите случаи на ОМАН (58), Griffin и съавт. (1996) описват клинични-
те и патохистологичните промени при ОМСАН (54). Освен с по-слабото
засягане на сетивните нервни влакна ОМАН се различава от ОМСАН и
по това, че болните показват по-бързо и по-добро възстановяване (16, 39,
40). Патохистологично при ОМСАН се установява периаксонално (адак-
сонално) разположени макрофагеални инфилтрати (подобно на ОМАН).
Освен периаксонално разположените макрофаги се наблюдават и оскъд-
ни лимфоцитни инфилтрати, както и незначителна демиелинизация. По-
добно на ОМАН и при ОМСАН се наблюдава елонгиране на нодалните
региони. И двете форми най-често последват предхождаща инфекция от
Campylobatter jejuni.
Фенотипно ОМАН и ОМСАН най-вероятно са две страни на патоге-
нетичен процес с еднаква локализация – имунна атака на аксона. Случа-
ите на ОМСАН с по-тежко засягане на вентралните коренчета (в сравне-
ние с дорзалните) подкрепя тази хипотеза, тъй като клиничната картина в
тези случаи е нещо средно между ОМАН и ОМСАН. Поради това Griffin
и съавт. (1996) смятат, че ОМСАН може да се разглежда като по-тежката
клинична манифестация на ОМАН (54).
Аксоналната увреда при ОМАН и ОМСАН най-често се дължи на
„молекулярна мимикрия“ на липополизахаридите от мембраната на
Campylobatter jejuni и някои от епитопите на нервните влакна. Това се
потвърждава от Capasso и съавт. (2011), провели серийни елекртродиаг-
ностични и имунологични изследвани при болни с ОМАН и ОМСАН,
при които невропатията е била предхождана от Campylobacter jejuni и
болните са били серопозитивни за GM1 и GD1a антитела (22). Невро-
физиологичните изследвания са показали засягане на сензорните влакна
не само при ОМСАН, но и в по-лека степен при ОМАН. И двете форми
се демонстрира обратима увреда на нервното провеждане както в мотор-
ните, така и на сензорните влакна. Освен това, не е възможно имуноло-
гично разграничаване на ОМАН от ОМСАН, тъй като титърът на IgG
aнтителата срещу GM1, GM1b и GD1a не се различава значимо при двете
форми (168).
III ЧАСТ
Хронична възпалителна
демиелинизираща невропатия
Хроничната възпалителна демиелинизираща полиневропатия
(ХВДП) e най-често срещаната хронична невропатия с болестност от
1 до 2 случая на 100 000 при възрастните (153, 167) и 0,5 на 100 000 в
детска възраст (47).
В 1958 г. Austin за първи път описва ХВДП при група болни с предим-
но двигателна невропатия, тежка мускулна слабост, показващи спонтан-
но или в отговор на стероиди клинично подобрение. Поради отсъствие
на мускулна атрофия Austin смята, че демиелинизация (а не аксонална
дегенерация) е причина за клиничната симптоматика (20).
Близо две десетилетия по-късно, в 1975 г., Dyck и съавт. публикуват
първото хистологично описание на ХВДП и предлагат наименованието
хронична възпалителна полирадикулопатия (69). Терминът демиелинизи-
раща е прибавен по-късно, когато ХВДП e дефиниран като самостоятел-
на нозологична единица. Хистологичното изследване е показало наличие
на сегментна демиелинизация, формиране на подобни на луковици уго-
лемявания, разположени периваскуларно в ендоневриума и епиневриума
мононуклеарни инфилтрати. Клиничната картина на изследваните болни
е двигателна полиневропатия с проксимална и дистална слабост и атак-
сична походка. Установен е бил повишен протеин в ликвора и неедно-
родно забавяне в проксималните сегменти на нервите. Въпреки близките
характеристики с острата възпалителна демиелинизираща невропатия,
според Dyck и съавт. (1975) клиничните, електрофизиологични и пато-
хистологични промени на описаните случаи са резултат от различно за-
боляване с хронично-прогресиращ или пристъпно-ремитентен ход (69).
1. Патохистологичните характеристики на ХВДП
Аутопсионните изследвания при ХВДП показват, че възпалителните
промени ангажират цялата дължина на нерва, но хистологичните про-
учвания локализират възпалителната патология основно в проксимални-
те нерви стволове, нервните коренчета и дисталните нервни сегменти
(94, 272).
Подобна предилекция на възпалителния процес е разбираема, поради
неефективната ендоневрална кръвно-нервна бариера в прооксималните
и дистални нервни участъци (201). Демиелинизиращите лезии засягат
както дългите, така и късите нерви, причина за сравнително равномер-
ното разпределение на мускулната слабост в ранната фаза на заболява-
нето. С напредване на болестния процес се обхващат и проксималните
стволове на нервите, като броят на демиелинизиращите лезии е по-голям
в по-дългите нерви, поради което мускулната слабост преобладава дис-
тално. Друга причина за конверсия на мускулната слабост от симетричен
в дистален патерн е последващата демиелинизиращото възпаление аксо-
нална дегенерация, чието протичане зависи от дължината на нерва.
Патохистологичните проучвания на спесимени от n. suralis показват
развитие на ендоневрален и периневрален оток. Възпалителните инфил-
трати са локализирани в епиневриума, периневриума и ендоневриума и
имат твърде малки размери или отсъстват (24, 70). Състоят се предимно
от макрофаги, CD3+ активирани Т клетки (основно CD8+ и CD4+ лим-
фоцити) и дендритни клетки (164). Подобни промени се наблюдават и
при други невропатии, поради което патогенетичната роля на клетъчните
инфилтрати е твърде съмнителна (48).
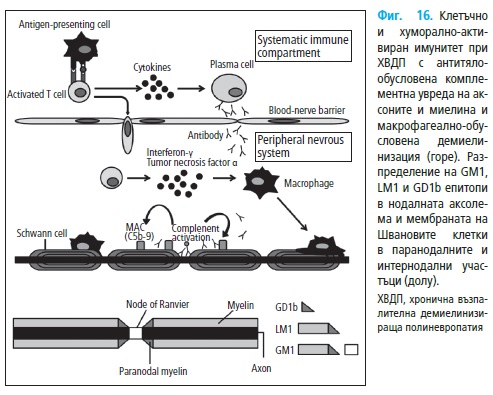
2. Имунопатогенеза на ХВДП
2.1. Клетъчен имуннен механизъм при ХВДП
В патогенезата на на ХВДП (Фиг. 16) основно е ангажиран клетъчни-
ят имунен отговор. Това се потвърждава от характеристиките на възпа-
лителните инфилтрати, промененото съотношение на T-клетъчните суб-
класове, увеличената експресия на цитокини и други проинфламаторни
медиатори, както и от участието на Т-клетките в патогенезата на експе-
рименталния алергичен неврит (ЕАН).
2.1.1. Роля на кръвно-нервната бариера
Увреждането на кръвно-нервната бариера има ключова роля в пато-
генезата на ХВДП. При физиологични условия кръвно-нервната бариера
подържа хомеостазата в ендоневриума, като предотвъртява свободното
движение на разтворими фактори (основно серумни протеини) от кръвта
към невралната микросреда.
Активацията на Т клетките увеличава пермеабилитета на бариера-
та за молекули, които иначе имат ограничен достъп до ендоневриума. В
активната фаза на заболяването CD4+ T клетките секретират проинфла-
маторни цитокини – интерлевкин (IL)-2 (102, 264), интеферон γ (IFNγ)
(155), а също така и хемокини – интерферон гама-индуциран протеин
(IP)-10 (132, 203) и макрофагеален инфламаторен протеин 3β (MIP3β)
(203). Освобождаването на цитокини и хемокини в циркулацията при-
чинява активация на макрофагите и индуцира свръхпроизводство в ен-
дотелните клетки на невралните микросъдове на адхезионни молекули,
каквито са васкуларната клетъчна адхезионна молекула VCAM-1 (15),
ендотелиалната левкоцитна адхезионна молекула ELAM-1 и интерцелу-
ларната адхезионна молекула ICAM-1 (178). Активираните Т-клетки се
прикрепят към ендотелните клетки чрез взаимодействие с адхезионни-
те молекули и се придвижват по повърхността на микросъдовете чрез
ротаторни движения, след което мигрират през бариерата. По време на
миграцията през микросъдовете Т-клетките продължават да отделят
възпалителни медиатори (матриксни металопротеинази и проинфлама-
торни цитокини/хемокини), което допринася за увеличението на пермеа-
билитета на нерва (102, 173).
Нарушената кръвно-нервната бариера е причина за наблюдаваното
при ХВДП МРТ контрастно (gadolinium) усилване на нервните стволове
(135).
2.1.2. Възпалителни инфилтрати
Биопсиите от n. suralis при ХВДП показват наличие на възпалителни
инфилтрати, съставени от CD8+ T-клетки (235), CD4+ T-клетки и макро-
фаги (48, 236). Локалната реактивация на Т-клетките, инфилтриращи
нервите, сe благоприятства от увеличената експресия върху мембранната
повърхност на макрофагите и Шванови клетки на антиген-представящи
комплекси на тъканна съвместимост (MHC) клас II (200) и костимули-
ращи B7-1/B7-2 молекули (130, 177). Проинфламаторните цитокини –
туморен некротизиращ фактор α и IFNγ IL-2 се продуцират от редица
имунни клетки (161) и активират имунния отговор.
От възпалителните клетки, инфилтриращи нерва, преобладават
макрофагите, формиращи клъстери около ендоневралните съдове (240).
Активираните макрофаги (локални и рекруитирани) играят ключова
роля за имунния отговор: антигенно представяне, освобождаване на про-
инфламаторни цитокини и токсични медиатори. Макрофагите играят
също така ключова роля в крайните стадии на демиелинизация, тъй като
фагоцититират разпадните миелинови продукти (130).
2.1.3. Роля на CD8+ T клетки
Ролята на CD8+ T-клетките в патогенезата на ХВДП е спорна.
В Швановите клетки се наблюдава хиперрегулация на MHC клас I
молекули, спомагащи за разпознаване и реактивация на цитотоксични-
те (CD8+) T-клетки (200). Реактивация на CD8+ клетки в ендоневриума
обаче се наблюдава не само при ХВДП, но и при лепроза, където инфек-
тираните от микобактерията Шванови клетки се лизират от специфични
CD8+ T-клетки (243).
На този етап при ХВДП не е открит нито чужд, нито собствен тарге-
тен CD8+ антиген, но ангигенно активиране е твърде възможно, тъй като
в засегнатите нерви е налице олигоклонална експанзия на CD8+ клетки
(235).
Олигоклоналната активация на Т клетките при ХВДП е по-скоро
маркер за хронична инфекция. На този етап in situ няма доказателства
за директен контакт между CD8+ и Шванови клетки, което значително
намалява вероятността CD8+ да играя роля на ефекторни цитотоксич-
ни клетки. Анализът на Т клетките показва по-манифестна активация на
CD8+ от тази на CD4+ T клетките, която постепенно намалява в процеса
на лечение с IVIg (165).
2.1.4. Т-регулаторни клетки и централен имунен толеранс
В здравите индивиди по-голяма част от автореактивните Т клетки се
елиминират след селекция в тимуса – централен имунен толеранс, но
някои успяват да избегнат елиминацията и попадат в периферната кръв.
По този начин има възможност за индуциране на автоимунен отговор. За
подържане на толеранса към собствени тъкани, автоимунните клетки са
подложени на постоянен контрол от супресивни Т регураторни клетки.
При ХВДП има индикации за нарушена имунорегулация, предпазва-
щата от ексцесивен и неадекватен имунен отговор (52, 230, 231). Броят
на циркулиращите Т регулаторни клетки при ХВДП е намален и ако бъ-
дат изолирани показват по-слаба от здравите контроли супресия на кле-
тъчната пролиферация (52, 230).
Променено взаимодействие между автореактивни Т клетки, анти-
ген-представящи клетки и възпалителни медиатори, водещо до спонтан-
но развитие на ХВДП, е наблюдаван в експременталния модел на не-
обезитусен диабет (NOD) при мишки с дефицит на костимулираща В7-2
молекула (228). Експерименталният модел е първоначално разработен,
за да се проучи ролята на Т клетъчната костимулация за развитие на заха-
рен диабет. Блокирането на B7-2 костимулация в тимуса предпазва миш-
ките от развитие на диабет, но неочаквано се развива автоимунна невро-
патия, клинично, електрофизиологично и хистологично съответстваща
на ХВДП. Хибридомното трансфериране показва, че невропатията се
медиира от миелин-P0-протеин-специфични CD4+ T клетки (150). Точ-
ковата мутация на автоимунно-регулиращия (Aire) ген води до намалена
експресия на P0 в тимуса, но в същото време настъпва увеличение на P0
специфичните T-клетки в периферията.
Подобно пренасочване на автоимунитета от панкреаса към пери-
ферните нерви е описан и при NOD мишки с дефицит на ICAM-1 (169).
Намалената експресия на ICAM-1 върху епителиалните клетки на тиму-
са променя Т клетъчната селекция и елиминация, като патогенетичният
процес се променя от диабетогенна в неврогенна посока (169).
2.1.5. Роля на макрофагите в имунопатогезата на ХВДП
Молекулярната мимикрия към епитопи на инфекциозни агенти е до-
казана при СГБ (290, 291).
Подобен механизъм се подозира, че би могъл да провокира патогене-
тична каскада при част от болните с ХВДП, особено тези с остра прогре-
сия, наподобяваща клиничната картина на СГБ (175).
Абнормното разпознаване на собствени миелинови епитопи от Т
лимфоцити, които обикновено се елиминират в тимуса, също би могло
да отключи автоимунния отговор при ХВДП (130).
Макрофагите играят ключова роля в патогенезата както на острите,
така и на хроничните възпалителни имунни невропатии (94).
Моноцитите, направлявани от адхезионните молекули селектин и
ICAM-1 (103, 256) и подпомагани от матриксините металопротеинази
навлизат от кръвта в ендоневриума и се диференцират в макрофаги. На-
чалният патогенетичен механизъм би могъл да се задейства и от резиди-
ращите в периферните нерви макрофаги, които изпълняват роля на анти-
ген-представящи клетки (130).
Свързването на антителата към нервните епитопи, последвано от
макрофагиална демиелинизация, е също възможен механизъм. При де-
миелинизиращата форма на СГБ с макрофагеално-индуцирана демие-
линизация е установено депозиране на компоненти на комплемента на
повърхността на Швановите клетки (99). Залавянето на антителата към
компонентите на миелина, освен че провокира комплементната каскада
(100), най-вероятно индуцира и директна макрофагеална фагоцитоза без
участие на комплемента (97). Проучванията при ХВДП обаче не нами-
рат подобно взаимодействие на антителата и макрофагите, но е доказана
директна увреда на миелина от макрофаги, секретиращи протеази (227).
Електронномикроскопските изследвания потвърждават активно-
то разграждане на иначе нормално изглеждащ миелин от активирани
макрофаги (204). Макрофагите проникват в нервите през ендоневрални-
те микросъдове (96), доближават се до миелиновите влакна и разпрости-
рат като „пипала“ цитоплазматичните си израстъци и пробиват базална-
та мембрана на миелиновите ламели. Следва инвазиране на вътрешното
спряму базалната мембрана пространство и стартиране на процеса на
разграждане на миелина. Вероятно протича и фагоцитоцитоза на миели-
нови компоненти, тъй като в цитоплазмата на макрофагите се наблюдават
движения от увредени миелинови ламели. Аксолемата остава незасегна-
та, дори когато макрофагите са в непосредствена близост. След приключ-
ване на миелиновото разграждане макрофагите, натоварени с миелинови
разградни продукти, излизат обратно навън, като в рамките на базалната
мембрана остават само демиелинизирани аксони (104). Швановите клет-
ки, локализирани в най-външните слоеве на миелина, изглеждат незасег-
нати от макрофагеалната фагоцитоза. Остатъци от тяхната цитоплазма,
разположени около демилинизираните аксони, персистират до момента,
в който стратира ремиелинизацията (201). Незасегнатите Шванови клет-
ки не само ремиелинизират оголените аксони, но също така и изместват
остатъците от цитоплазма на засегнатите Шванови клетки (134). В ре-
зултат от това се оформят уникални, подобни на луковици удебеления,
които обграждат аксона (134, 201).
Подобен процес на макрофагеално обусловена демиелинизация/ре-
миелинизация е докладвана и при EАН (237).
Лонгитодинални спесимени от n. suralis показват, че макрофагите за-
сягат както нодуса, така и интернодалните региони на засегнатите нерви
(104). В повече от половината случаи макрофагеалните лезии се локали-
зират в интернодуса. Последният лесно се разпознава по инцизурите на
Schmidt-Lanterman, където се наблюдава фокално разслояване на миели-
новите ламели (104).
Нодалните региони са друга възможна локализация на макрофагеал-
но- обусловената демиелинизация. Наблюдава се придърпване встрани
на паранодалните ламели, като израстъците на макрофагите се разпола-
гат между миелина и аксолемата. Макрофагите по-често инвазират само
един от двата съседни паранодуса, но е възможно и засягане в двете по-
соки (104). При нодална инвазия, макрофагите първо разрушават миели-
на на паранодуса, след което се насочват в посока към интернодуса (226).
Специфичните места на макрофагеално-обусловена деструкция на
миелина показват, че макрофагите разпознават специфични епитопи по
тяхната повърхност (104).
2.2. Хуморални механизми
Ефектът от плазмаферезата показва, че хуморалният имунитет също
играе важна роля в патогенезата при ХВДП (Фиг. 16).
Патохистологичните и серологични проучвания предоставят неопро-
вержими доказателства за активиране на хуморалния имунен механизъм
(110).
Имунохистохимичните проучвания установяват отлагане на имуног-
лобулин и съставки на комплемента по външната повърхността на Шва-
новите клетки при иначе компактен миелин (55, 107). При индиректна
имунофлуоресценция е доказано, че серумът на пациентите с ХВДП се
свързва към епитопите на нормални нерви (284). Интраневралното ин-
жектиране на серум от болни причинява демиелинизация (284), като тар-
гетен антиген е идентифициран компактния миелинов P0 протеин (285).
При повечето случаи на ХВДП специфичният таргет на автоимун-
ната атака остава неизвестен, но хистологичните промени в миелина
предполагат ангажиране на някои от протеиновите му компоненти. Ин-
дуцираният от P0 (171), P2 (122) и периферен (PMP)-22 (85) миелинов
протеин EAN, показва че автоимунният отговор срещу тези детерминан-
ти има потенциал за индуциране на заболяване с увреждане на перифер-
ните нерви. Автоимунен имунен отговор е докладван и срещу P2 (115),
PMP-22 (85) и конексин (141).
Проучването на Sanvito и съавт. (2009) обаче отхвърля възможността
за индуциране на антитяло-обусловен автоимунен отговор (231).
3. Клинични характеристики на ХВДП
Болните с ХВДП първоначално се манифестират с тръпнене и мравуч-
кане, които скоро след това се последват от постепенно влошаваща се
мускулна слабост. Засягат се както дисталните (малки мускули на ръце-
те, екстензионна пареза на ходилата), така и проксималните мускули (за-
труднение при изправяне от седнало положение, издигане на обекти над
главата). В повечето случаи най-проблематичен е моторният дефицит.
Пациентите имат затруднения при ходене и чести падания, което налага
използването на помощни средства (дори инвалидна количка).
За разлика от честите оплаквания от намалена сетивност и сензорна-
та атаксия (болните нямат усет за положението на крайниците в прос-
транството), автономни симптоми и невропатична болка се наблюдават
по изключение. Когато болката е налична, тя не е основен симптом и
само в одлени случаи може да е силна.
При ХВДП отделните популации нервни влакна се засягат нееднак-
во, като демиелинизацията преференциално уврежда дебелите нервни
влакна (моторни и проприоцептивни), докато тези с тънък миелин или
без миелин (болкови и автономни) обикновено остават съхранени.
Пациентите са увредени в различна степен; от минимално (остават
амбулаторни) до тежко засягане (на инвалидна колична до невъзможност
да се хранят и обслужват самостоятелно). Дихателната мускулатура е
рядко ангажирана от болестния процес и не се налага използване на апа-
рати за апаратна вентилация. Засягането на краниалните нерви е изклю-
чение, като най-често се увреждат околомоторния и лицевия нерв.
Разграничаването на ХВДП и ОВДП се основава на различния вре-
меви ход. При ОВДП симптомите се развиват бързо в рамките на дни до
седмици, като максималният дефицит се достига в рамките на 1 месец.
След неговото достигане мускулната сила се подобрява постепенно, въ-
преки че остатъчен дефицит не е изключен. Противоположно на ОВДП
симптомите и неврологичният дефицит при ХВДП се развиват бавно в
продължение на месеци (най-малко 2 месеца). Бърз пристъпно-реми-
тентен ход (подобен на ОВДП) е също възможен, но честото повтаряне
на пристъпите и персистирането на неврологична симптоматика в меж-
дупристъпния период e по-типично за ХВДП. Пристъпно-ремитентният
ход се среща при деца и млади възрастни и се разграничава от ОВДП
по по-високата честота на обострянията при ХВДП. Въпреки че перси-
стирането на неврологичия дефицит в междупристъпния период гово-
ри в полза на ХВДП, резидуален дефицит може да се наблюдава и при
ОВДП. Влошаването на неврологичния статус във времето е в подкрепа
на ХВДП.
За разлика от ОВДП, който в повечето случаи е постинфекциозен не-
врит, при ХВДП обикновено не се регистрират предхождащи инфекции.
Причината за това може да се дължи на постепенното и незабележимо
начало, при което болните не винаги могат определят момента на възник-
ване на симптомите.
При неврологичното изследване се открива моторен дефицит със
слабост в раменни и тазобедрени мускули, а също така в ръцете и хо-
дилата. Засягането е обикновено симетрично и показва тенденция за
обхващане както на долните, така и на горните крайници. Въпреки ед-
новременното ангажиране на проксималните и дистални мускулни гру-
пи, клиничният дефицит е по-манифестен дистално. При изследване на
сензорните нерви се установява по-тежко засягане на проприоцептивен
и вибрационен усет (миелинизирани влакна с голям диаметър). Дълбо-
ките сухожилни и надкостни рефлекси обикновено не се получават или
са дифузно отслабени.
Съгласно критериите на American Academy of Neurology (AAN)
клиничната картина на ХВДП се характеризира с „моторна и сензор-
на дисфункция на повече от един крайник“. Според тeзи критерии се
включват различни фенотипни варианти, в това число мултифокалната
придобита демиелинизираща сензорна и моторна невропатия (МПДСМ)
и мултифокалната моторна невропатия (232).
В 2005 г. Joint Task Force на European Federation of Neurological
Societies и Peripheral Nerve Society (EFNS⁄PNS) класифицират ХВДП в
различни клинични субтипове. Kласическият ХВДП се класифицира
като „типичен“ ХВДП, а към „атипичния ХВДП“ се отнасят МПДСМ
(асиметричен ХВДП) и дисталната придобита симетрична невропатия
(ДПДС). Съгласно критериите на EFNS⁄PNS от общата група на ХВДП
се изключват мултифокалната моторна невропатия (MMН) и анти-MAG
гликопротеиновата невропатия.
IV ЧАСТ
Концепцията нодо-паранодопатия
Нодо-паранодалните региони на дисталните аксонални терминали
са своеобразно „бойно поле“, на което се разиграва имунната атака при
аксонални форми на синдрома на Guillain–Barré (СГБ), мултифокална
моторна невропатия (ММН) и по-рядко при хронична възпалителна де-
миелинизираща невропатия (ХВДП).
В нодалния регион са локализирани ганглиозиди, гликосфинголи-
пиди, дизиалозилови гликолипиди и насочените срещу тях антитела,
които се асоциират с аксонални форми на (19).
При ХВДП е ангажиран основно паранодалния регион, където тар-
гет на имунната атака са адхезионните протеинови молекули.
1. Нодопатии
1.1. Антиганглиозидни антитела
Ганглиозидите са широко разпространени в клетъчните мембрани на
периферната и централна нервна система. Тяхното количество варира в
моторните, сензорни (9) и краниални нерви (44). Таргети на антитяло-
вата атака са ганглиозидни епитопи, локализирани в нодуса – GM1 (20),
GD1a (26) и GD1b (8) и паранодалния регион (43).
Клиничният фенотип корелира не само с локализацията (28), но
зависи от специфичните характеристики на отделните субкласове ан-
титела. Антителата срещу GM ганглиозиди, експресирани в нодалната
аксолема и микровилите на Швановите клетки, най-често се асоциират
с остра моторна аксонална невропатия (ОМАН) (IgG клас) (20) и MMН
(IgM клас) (16). Механизмът, по който антителата предизвикват чисто
моторен фенотип невропатия не е напълно изяснен, тъй като ганглиозид-
ните епитопи са сравнително еднакво разпространени в моторните и се-
тивни нерви (9). Способността на антителата срещу GM1 да разпознават
1.2. Антитела срещу нодални протеини
Експерименталният алергичен неврит е модел на демиелинизираща-
та форма на СГБ. Като имуноген се използва периферен миелин, чиито
патологичен ефект се свързва с деструкция на адхезионните молекули
на нода и последваща паранодална „демиелинизация“. Тeзи промени на-
стъпват в резултат от синтеза на антитела срещу NF186 (неврофасцин) и
GLDN (глиален протеин), които нарушават клъстера на Na+ каналчета и
последващо увреждане на нервното провеждане (29).
Антителата срещу NF186 се асоциират с ОМАН, докато тези сре-
щу GLDN са по-чести при ОВДП. Повече от половината СГБ болни са
негативни за тези антитела, но това не изключва съществуването на ан-
титела срещу други епитопи, които на този етап остават неидентифици-
рани. Серумите на някои от болните с ОМАН съдържат антитела срещу
адхезионни молекули (NF186, GLDN or CNTN), като едновременно с
това са позитивни и за IgG-GM1 антитела, тъй като патологичният про-
цес тригерира синтез на повече от един вид антитяло (29).
В експерименталните модели, медиирани от пасивен трансфер на
GLDN антитела, се наблюдава ранна нодална увреда и паранодална
„демиелинизация“ с депозиране на IgG и активация на комплемента.
По-късно възникващата демиелинизация се асоциира с инфилтрация на
T-клетки и макрофаги. Пасивният трансфер на анти-GLDN IgG и P2 ан-
титела увеличава честотата на демиелинизиращите лезии, но не предиз-
виква аксонална дегенерация (30).
V ЧАСТ
Вместо епилог: електродиагностични проблеми, нови диагностични и
терапевтични стратегии
1. Електродиагностични проблеми при Guillain–Barré
синдром
На основа на клинични, електрофизиологични и патологични харак-
теристики на Guillain–Barré (СГБ) се класифицира в две (засега) основ-
ни форми: демиелинизираща и аксонална (24, 49). В Северна Америка и
Европа (12, 42) преобладава демиелинизиращата, докато в Китай (49) и
Япония (69, 89) аксоналната форма.
Неопровержим факт е липсата на „златен диагностичен стан-
дарт“ за СГБ (127). След 1990 г. електродиагностиката на СГБ стана още
по-комплицирана, тъй като освен острата възпалителна демиелинизира-
ща полирадикулоневропатия (ОВДП) се описаха две аксонални форми
– моторна аксонална невропатия (ОМАН) и остра моторна и сензорна ак-
сонална невропатия (ОМСАН). Обикновено аксоналните форми се пред-
хождат от Campylobacter jejuni инфекция и се асоциират с продукцията
на антиганглиозидни антитела. Въпреки аксоналния характер на увре-
дата, някои от болните с ОМАН се възстановяват по-бързо и по-добре от
тези с ОВДП (50, 68).
За диференциране на ОВДП и ОМАН в продължение на две десети-
летия се използваха диагностични групи критерии на Ho и съавт. (1995)
(49) и Hadden и съавт. (1998) (42).
Съгласно критериите на Ho и съавт. (1995) СГБ се класифицира
като ОВДП при установяване на сигнификантно забавяне на провежда-
нето по нервите: дистална моторна латенция (ДМЛ) > 110% от горната
граница, моторна скорост (MC) < 90% от долната граница на нормата, F
латенция >120% от горната граница или > 50% блок в провеждането (ПБ)
или „несъмнена“ (абнормна, ексцесивна) темпорална дисперсия (TД) в
поне два моторни нерва.
Критериите на Hadden и съавт. (1998) се различават от тези на Ho
и съавт. (1995 по това, че не включват „несъмнената“ TД. Първона-
чално се считаше, че ОМАН се причинява единствено от аксонална де-
генерация, поради което електрофизиологичните критерии включваха
единствено редуцирани по амплитуда моторни отговори и отсъствие
на белези на сигнификантна демиелинизация (42, 49).
Очакваше се описаните критерии да подобрят диагностичните въз-
можности на неврографията при класифициране формите на СГБ. Въз-
никна обаче нов проблем.
В 1998 г. Kuwabara и съавт. описаха при позитивните IgG анти-GM1
СГБ пациенти един електродиагностичн феномен, наречен обратим ак-
сонален блок (RF, reversible conduction failure) (68). Проводният блок
е най-вероятно имуномедииран от антитела (51, 89), играещи патогене-
тична роля за блокиране на Na+каналчета (49, 69, 137). При 10 – 42% от
случаите при СГБ се установяват високи титри на анти-GM1 антитела
(34, 49, 106), чиято патогенетична роля за аксоналната увреда не е докрай
изяснена (45, 47). Пасивният трансфер на GM1 антитела в експеримен-
тални модели предизвиква проводен блок, който се съпровожда от реду-
кция на Na+ каналчета (11, 109, 138, 139).
Неврографиите в случаи с RF демонстрират възстановяване на пър-
воначално редуцирани или липсващи моторни отговори (или сигнифи-
кантно забавеното провеждане) до нормални стойности (18, 69, 125).
Това създава допълнителни затруднения, тъй като част от електродиаг-
ностицираните ОВДП форми, поради „демиeлинизиращо“ забавяне и/
или блок, са всъщност аксонални форми на СГБ с RF (22, 46, 64, 65, 113,
123, 125, 126).
Оформиха се два подхода за решаване на този диагностичен проблем
(127).
Първият подход разчита на т.нар. серийни (повторни) неврографии
(СН), които при ОМАН показват бързо възстановяване на проводния
блок в терминалните окончания (69) и в интермедиерните сегменти (70)
на засегнатите нерви. На основа на този подход в 4 отделни проучвания
(211 пациенти със СГБ) е установено, че 22 – 38% от GBS случаи проме-
нят своята класификация от ОВДП (или некласифицируеми) в аксонална
форама на СГБ (64, 65, 113, 125).
Друга група изследователи имат противоположно становище. Спо-
ред не е необходимо провеждане на СН, при условие че се използват
по-строги мозифицирани критерии за демиелинизация (53, 104, 132).
Van den Bergh и Piéret (2004) предлагат по-стриктни изисквания за де-
миелинизация и блок: редуцирана моторна скорост < 70%, дистална мо-
торна латенция > 150%, F-латенция > 120% или > 150% (ако дисталния
CMAП е < 50%), проксимален CMAП/дистален CMAП отношение < 0,7
(> 70%) (128).
Ретроспективното проучване на Rajabally и съавт. (2015) (104) и
проспективното проучване на Van den Bergh и съавт. (2018) (132) показ-
ват, че класификацията на СГБ не се променя съществено при условие, че
се прилагат модифицираните критерии, което обезмисля провеждането
на СН.
Привържниците на СН подход (64, 113, 125) отстояват тяхната ди-
агностична роля, тъй като единствено повторните неврографии могат
дават оценка на цялостната електродиагностична картина, отразя-
вайки евентуалното влошаване на демиелинизацията, появата на реми-
елинизиращи белези и най-вече протичането на RCF. Според Uncini и
съавт. (2010) критериите за RCF трябва да включват > 150% увеличение
на дисталната CMAП амплитуда (при CMAП продължителност < 120%)
или редукция на проксималния блок с > 0,2 (при проксимална CMAП/
дистална CMAП продължителност < 130%) (125).
Противоречивите еспертни становища възпрепятстват единствено
електродиагностична класификация на СГБ, тъй като на този етап аксо-
налните и демиелинизиращи форми имат идентична терапия.
Разработването на нови терапевтични стратегии обаче налага разре-
шаването на този проблем, като едно евентулно проспективно сравнение
на едни и същи електродиагностични показатели при двата подхода със
задължително отчитане на RCF изглежда разумно решение.
2. Електродиагностични проблеми при ХВДП
Хроничната възпалителна демиелинизираща полиневропатия
(ХВДП) е лечимо имуномедиирано заболяване, засягащо 1,0 – 8,9 болни
на 100 000 души (80). Навременното диагностициране и лечение са от
особена важност за предотвъртяване на развитието на необратим невро-
логичен дефицит (15).
Изненадващи и обезпокояващи са публикуваните наскоро данни, че
близо половината (47%) от болните диагностицирани и лекувани за
ХВДП не покриват минималните диагностични критерии. От дру-
га страна, вероятността за диагностична грешка и неподходящо лечение
значително нарастват поради съществуването на т.нар. атипичните фор-
ми на ХВДП (8).
До този момент има повече от 15 групи диагностични групи крите-
рии, но не е известно колко от тях се прилагат в ежедневната практика (2,
16). Диагностиката на ХВДП разчита основно на електродиагностичното
документиране на демиелинизация, често пъти без да се базира на спе-
цифичен протокол (1).
Критериите за ХВДП се основават на експертен консенсус (130). Раз-
работени са протоколи както за типичните, така и за атипичните вариан-
ти, като EFNS/PNS критерии имат най-висока диагностична чувст-
вителност (15). Диагнозата ХВДП се приема, ако типичните клинични
белези (проксимална и дистална мускулна слабост) се съпровождат от
демиелинизиращи промени в най-малко 2 периферни нерва (73% чувст-
вителност и 88% специфичност). Ако критериите за демиелинизация се
покриват само от 1 нерв или електродиагностичните промени не дости-
гат сигнификантни нива, промените в ликвора, MРТ неврография и нерв-
ната биопсия имат подкрепяща роля и могат да подобрят диагностична
стойност до 91%, но в някои случаи я редуцират до 66% (15).
От това следва, че клиницистите трябва да подлагат на критична
оценка находките от електродиагностичното изследване, които трябва
да се разглеждат в контекста на клиничната находка.
Това дава основание за търсене на нови диагностични техники и
стратегии.
3. Нови диагностични техники
3.1. Напреднали електродиатностични техники
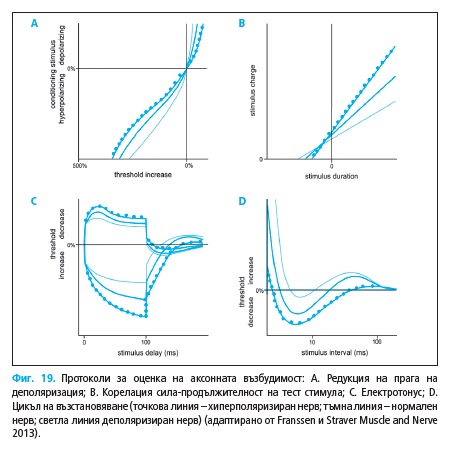
3.1.1. Оценка на аксонна възбудимост
В 1998 г. в Института по Неврология в Лондон Bostock разработва
неинвазивна неврофизиологична техника за оценка прага на мембранна-
та деполяризация (14). За целта е използвана софтуерна QTRAC 4.3 вер-
сия (14), която по-късно е заменена от разработените от Kiernan и съавт.
(2000) TRONDHM протоколи (61).
Прави се оценка на биофизичните характеристики на мембрана в
местата на стимулация, осигуряваща директна информация за функци-
ята на Na+ и K+ каналчета (14). Проследяването на деполяризационния
праг е възможно единствено ако провеждането на нервният импулс меж-
ду мястото на стимулация и отвеждането от мускула е запазено.
Основната характеристика, която се оценява, е силата на тест сти-
мулите (праг на деполяризация), необходима за постигане на сума-
рен акционен потенциал с определен размер, като таргетът е 30-40% от
максималния сумарен потенциал (60, 61). Изследването е безболезнено.
Оценява се възбудимостта на моторните аксони на n. medianus след сти-
мулация в областта на китката и отвеждане от abductor pollicis brevis.
Стимулацията се осъществява и контролира от компютър. Катодът се
поставя върху китката, а анодът се позиционира 10 cm проксимално.
Амплитудата на CMAП се записва от abductor pollicis brevis с активен
електрод върху моторната точка и референтен, поставен върху прокси-
малната фаланга. Регулярно се подават два типа стимули: тест стимули
с продължителност от 0,2 или 1 ms (на интервали от 0,8 sec) и надпрагови
или субпрагови (поляризиращи или хиперпоряризиращи) кондиционира-
щи стимули. Кожната температура се подържа над 32°C (62).
Прагът на деполяризация може да бъде оценен на основата на ня-
колко протокола.
Сила-продължителност константа τSD e нодална характеристика
(Фиг. 19B), отразяваща корелацията на намалението на прага на деполя-
ризация от увеличението на продължителността на стимула.
τSD = 0,2(I0,2 – I1,0)/(I1,0 – 0,2 I0,2)
τSD – константа сила-продължителност
I0,2 и I1,0 – сила на прагов ток при продължителност на тест стимулите
съответно 0,2 и 1,0 ms
Реобазата представлява силата на праговия стимул за достигане на
т.нар. „безкрайна“ дължина на стимула, чието надвишаване не води до
намаление на праговия ток, а се последва от отсъствие на отговор. Реоба-
зата е обратно пропорционална на τSD константа:
Irh = t x I/(t + τSD)
Irh е сила на тока при достигане на реобазата; t – продължителността на
стимула; τSD – константа сила-проължинелност; I – сила на тока при
тест стимули с τ продъжителност
Цикълът на възстановяване отразява възстановяването на аксонната
възбудимост при подаване на двойка стимули. Подават се супрамакси-
мални кондициониращи стимули и интервали от 2 – 200 ms тест стимули
с продължителност от 1,0 ms. Регистрират се единствено отговорите на
тест стимулите, получени след субтракция на предизвиканите от конди-
циониращата стимулация отговори (Фиг. 19D).
При изследване на праговия електротонус се прави оценка на ефек-
та на променения мембранния потенциал върху прага на деполяриза-
ция. Промяната на мембранният потенциал се постига чрез подаване на
субпрагови (40% от прага) деполяризиращи и хиперполяризиращи кон-
дициониращи импулси с продължителност от 100 ms, комбинирани със
серия от тест стимули с къса продължителност (Фиг. 19C).
Редукцията на прага за деполяризация се изследва чрез подаване
субпрагов кондициониращ (поляризиращ или хиперполяризиращ) им-
пулс с продължителност от 200 ms и след неговото приключване се ос-
вобождава тест стимула. Силата на кондициониращия ток се променя от
50% (при деполяризиращия) до 100% (при хиперполяризация) и графич-
но се оценява корелацията на тази промяна с прага за постигане на тар-
гетния отговор (Фиг. 19A).
Kuwabara и съавт. (2002) използват техника за изследване на ак-
сонната възбудимост за диференциране на ОМАН от ОВДП (70). При
ОМАН се наблюдава манифестно увеличена рефрактерност, последвана
от бърза нормализация и възстановяване на нормалните характеристи-
ки на мембраната (Фиг. 20A). Аксонната възбудимост при ОВДП не се
съпровожда от манифестни промени след стимулация в областта на кит-
ката (Фиг. 20B). Това най-вероятно се дължи на факта, че патологичните
промени при ОВДП са по-манифестни в разположените дистално от кит-
ката нервни терминали. Увеличена рефрактерност не се наблюдава при
амиотрофична латерална склероза и диабетна полиневропатия, тъй като
мембранните промени при ОМАН вероятно не се дължат на аксонална
дегенерация, а на удължаване на рефрактерния период, при което се бло-
кира втория от двойката тест стимули (71).
При пациентите с ХВДП n.medianus в областта на китката е засегнат
предимно от демиелинизация. Сегментната демиелинизация води до на-
маление на съпротивлението на миелиновата обвивка (7). Най-изразени
промени на аксонната възбудимост се наблюдават при изследване на пра-
говия електротонус (Фиг. 21). Наблюдава се „разперване“ на проговия
електротонус при 48% от болните (19, 118), което корелира с продъл-
жителността и тежестта на заболяването (118). Променените индекси на
аксонната възбудимост се нормализират след иницииране на лечение с
IVIg (75).
Въпреки продължаващите дебати за патогенезата на MMН и по-спе-
циално дали промените на нервната проводимост се дължат на демиели-
низиращ или аксонален процес, неврографски MMН се характеризира
като генерализиран процес с фокално представяне, при който не се ус-
тановяват промени извън местата на блока (20). Аксонната възбудимост
дистално от местата на проводния блок обаче установяват сигнификант-
ни промени (36, 63). Наблюдава се сигнификантна промени в наклона
на кривата при изследване на редукцията на прага на деполяризация,
разперване на праговия електротонус и увеличение на периода на су-
первъзбудимост при оценка на цикъла на възстановяване при подаване
на двойка стимули (Фиг. 22). Тези промени на аксонната възбудимост,
изключително абнормни при MMН, зависят от проводните специфики
на паранодалната и интернодална аксонална мембрана, като участъкът
от нерва, разположен дистално от мястото на проводния блок показва
типични за хиперполяризация характеристики, наподобяващи пост-исхе-
мични промени.
3.1.2. Оценка на моторната скорост на провеждане чрез SF-EMG
електроди
CMAП представлява сумарна активност, генерирана от моторните
единици в рамките на мускула, след стимулация на нерва. Размерът и
формата на CMAП зависят от потенциалите на индивидуалните моторни
единици, техния брой и позициониране в мускула, скоростите на про-
веждане на отделните моторни влакна, както и от дължината между мяс-
тото на стимулация и отвеждане от мускула.
Скоростта на провеждане (CП) на нормалните аксони е пропорцио-
нална на техният диаметър (107, 137). CП зависи също така и от мем-
бранните характеристики, температурата и състоянието на миелиновата
обвивка. Изследването на CП е важно от клинична гледна точка, тъй като
при миелинопатия тя е редуцирана (55, 83).
Най-често CП се определя с повърхностни електроди – конвенцио-
нална скорост на провеждане (C-CV). C-CV обаче не отразява опти-
мално проводимостта по периферните нерви, тъй като отчита единствено
провеждането по бързопровеждащите нервни влакна на алфа моторната
популация (10). При много от патологичните процеси нервните влакна
не се засягат еднакво (73, 83). Промените на бавнопровеждащата група
аксони може да бъде оценена индиректно и само частично на основа на
C-CV. Така напр. C-CV може да е в нормални граници дори когато ам-
плитудата на CMAП е редуцирана, а отговорът десинхронизиран (91).
Това значително намалява чувствителността на C-CV при оценка на не-
вропатии със засягане предимно на бавнопровеждащи нервни влакна и
при невропатии, при които нервните влакна не са еднакво ангажирани,
каквито са възпалителните демиелинизиращи невропатии, при които
лезиите са пръснати по хода на нерва.
Single Fiber (SF)-EMG се провежда чрез специално конструирани иг-
лени електроди, при което има възможност за отвеждане на потенциали
от индивидуални мускулни влакна (6). По-големите възможностите на
SF-EMG техники се обуславят от малката записваща повърхност на иг-
лите с диаметър от 25 μ (117). Електродиагностичната чувствителност
на SF-EMG за диагностика на проводния блок е висока, тъй като предос-
тавя възможност за оценка на проводимостта на индивидуалните мотор-
ни единици (92, 93). При тази техника отвеждането става от произволни
места в мускула, което дава възможност за изследване на различни групи
индивидуални моторни аксони.
Определянето на скоростта от единични влакна (SF-CV) се раз-
личава от C-CV единствено по това, че вместо повърхностни електроди
се използват SF-EMG електроди. SF-EMG игла се поставя в мускула и
нервът се стимулира супрамаксимално в две различни места – дистално
и проксимално (подобно на C-CV). CMAП сe отвежда първо с конвен-
ционални повърхностни електроди и се определя супрамаксималната
стимулация. След това интензивността на стимула се увеличава с 15% и
се записва максималната CMAП амплитуда. Филтрите са фиксирани на
500 Hz (високочестотен) и 10,000 Hz (нискочестотен).
Чрез SF-EMG електроди се записват потенциалите, получаващи се в
резултат от супрамаксимална дистална стимулация на нерва. Критери-
ите за това кои от потенциалите са годни за анализ е тяхната форма, като
се анализират само острите, спайкови отговори с късо време за издигане.
Прилага се втората (супрамаксимална) стимулация, която трябва да про-
дуцира еднакъв на първия по амплитуда и форма отговор, което е гаран-
ция, че позицията на иглата остава непроменена. Ако вторият отговор е
различен от първия, позицията на иглата се променя и цялата процедура
се повтаря отново.
Същата техника се прилага и проксимално. Отговорите, получени при
проксимална и дистална стимулация, трябва да имат една и съща форма.
Обикновено са необходими около 13 опита за получаване на 10 отвеж-
дания, без да има нужда да се променя положението на иглата. Според
авторите на оригиналното описание (92, 93) тази процедура не е особено
трудна за квалифицирани неврофизиолози и не налага особен опит.
SF-CV се изчислява според началото на девиацията от основната ли-
ния, въпреки че могат да бъдат използвани и пиковете на отговорите.
За всеки нерв са необходими 10 SF-CV, регистрирани при произволно
преместване на SF-EMG електрод. Прието е 36 m/s да се счита за долна
граница на нормалната SF-CV, тъй като в литературата тези стойности
се приемат за нормални стойности на най-бавно провеждащите алфа мо-
торни аксони (25, 29, 56, 121, 128) и следователно стойности под 36 m/s
са белег на демиелинизация.
Когато има съмнение за засягане на дисталните нервни сегменти
може да се анализира дисталната моторна латенция. В тези случаи
нормалните граници на отговорите, получени при използване на SFEMG
eлектроди, се получават на основа на стойностите, получени с по-
върхностни електроди при здрави индивиди плюс 30%.
SF-EMG техника може да се използва за изследване на скоростите
по n. fibularis, n. tibialis, n. ulnaris. Отвеждат се съответно отговорите от
extensor digitorum brevis при стимулация в областта на глезена и фибу-
ларната глава; abductor hallucis при стимулация в медиалния глезен и по-
плитеалната ямка; abductor digiti minimi при стимулация в китката и под
лакътя (92, 93).
Когато са засегнати едновременно тънките и дебели аксони C-CV е
патологично променена, само при тежка миелинова увреда на дебели-
те нервни влакна (17). Това е така, защото конвенционалната неврогра-
фия зависи основно от бързопровеждащите аксони. При увреждане на
малък брой аксони C-CV остава непроменена. При SF-CV нервните
влакна се изследват едно по едно, което значително повишава диагнос-
тичната чувствителност. При съмнение за проводен блок описаната тех-
ника може да се използва за оценка на отделните сегменти на нерва (92,
93). Произволното записване от различни мускулни влакна (премествай-
ки електрода в мускула) позволява оценка на ралични групи от аксони.
Borg и съавт. 1979 са описали подобна техника, но с концентрични игле-
ни електроди (13).
SF-EMG електроди осигуряват по-голяма селективност, като регис-
трират CП от малка група аксони (или от единичен аксон). Директно-
то сравняване на SF-CV и C-CV в една и съща група демиелинизиращи
невропатии показва, че определената със SF-EMG електроди скорост
има по-висока чувствителност, като се регистрират абнормна SF-CV в
повече от половината нерви с нормална C-CV. Небходими са обаче про-
учвания и при компресивни невропатии, както и при други патологични
състояния, за да се разбере дали редукцията на SF-CV се дължи един-
ствено на демиелинизация или може да отразява и друг тип патология
(напр. дегенерация-регенерация или аксонална увреда).
3.2. Магнитно-резонансна неврография и ултразвуково
изследване на периферни нерви
При пациенти с хронични имуномедиирани невропатии възпаление-
то и миелиновата дисфункция водят до задебеление на нервите, което
може да се визуализира при магнитно-резонансна томография (MРТ) и
ултразвуково изследване (30, 67, 119).
MРТ неврография се използва основно за изследване на промените
в спиналните коренчета, докато ултразвуковото изследване се фоку-
сира основно върху периферните нерви на горни и долни крайници (35,
37, 38, 133).
Важен недостатък на изобразителните методи на нервите при хро-
нични имуномедиирани невропатия е отсъствието на системен анализ
на диагностичната стойност. Прилагат се различни протоколи, което
затруднява интерпретацията и сравнение на находките.
Независимо от тези недостатъци изобразителните изследвания при
хронични имуномедиирани невропатии несъмнено могат да визуализи-
рат удебеления по дължината на нервите, чиито локализации често се
разминават от електродиагностично установения блок на провеждането.
Следваща стъпка е използването на различни протоколи за определяне
патерна на удебеляване на периферните нерви. Анализират се промени-
те в структурата (нормална структура, леко, фокално и дифузно удебе-
ляване), анатомичните места и наличието на фасцикуларно засягане (40,
41, 94, 95, 57-59, 142).
MРТ на плексуса разчита основно на T2-секвенции (DIXON или 2D
кратковременно tau инверзионно възстановяване – STIR) и може да де-
монстрира удебеляване и/или хиперинтензен сигнал в нервните коренче-
та, както и постконтрастно (gadolinium) усилване (133). В малък брой
ретроспективни проучвания с ограничен брой участници е изследвано
разпределението на промените в плексуса (нормално, симетрично или
фокално, предимно в коренчетата или фузиформено) (54, 103, 116, 131).
При ХВДП и мултифокална моторна невропатия (MMН) MRI промените
показват значими вариации (40 – 100%). Най-често срещаните промени
са наличието на хиперинтензни сигнали (44 – 72% при ХВДП; 44 – 100%
при MMН), следвани от удебеляване на нервите и постконтрастно усил-
ване (30, 67). Симетрични MРТ промени в плексуса се асоциират с гене-
рализирана слабост, докато фокалните абнормности с асиметрична та-
кава, но корелациите не са потвърдени в проспективни проучвания (54).
Разработени са протоколи за количествена MРТ оценка на промените в
брахиалния плексус, значително увеличаващи диагностичната чувстви-
телността при MMН, Lewis-Sumner синдром (LSS) и ХВДП (102).
MРТ неврография на брахиалния плексус трябва да се интерпрети-
ра в контекста на клиничните промени, тъй като освен ХВДП няколко
други невропати – Charcot-Marie-Tooth (CMT) тип 1, невропатии с IgM
гамопатия, диабетна плексо-радикуло-невропатия, невролимфоматоза,
невралгична амиотрофия, васкулитни синдроми и неврофиброматоза де-
мострират сходни промени (102).
В последните години се разработиха протоколи за количествена
оценка на промените при ултразвуково изследване на периферните нер-
ви, както и специализирани такива за ХВДП и MMН (39). Удебеляване-
то на n. medianus в областта на предмишницата и мишницата в ком-
бинация с удебеляване на някой от стволовете на брахиалния плексус
има 99% специфичност за възпалителна невропатия. Установяването на
2 абнормни нервни сегмента при двустранно ултразвуково изследване
на n. medianus в комбинация със засягане на брахиалния плексус също
има висока специфичност. При отсъствие на клинични белези за деми-
елинизираща наследствена невропатия, ултразвуковото изследване на
проксималните участъци на n. medianus може успешно диагностицира
ХВДП, LSS и MMН (39). Протоколът за ултразвуково изследване на пе-
риферните нерви може да се съкрати в ежедневната практика без това да
намали диагностичната им стойност. Размерът на периферните нерви
е най-важният сонографски параметър при възпалителни невропатии,
докато фасцикуларният размер, ехогенността и васкуларизацията нямат
допълнителна диагностична стойност.
Не е известно дали резултатите, получени при MРТ неврография и
ултразвуковото изследване са диагностично сравними при ХВДП (76,
136), въпреки че комбинирането на двете техники увеличава диагнос-
тичната чувствителност при ХВДП и MMН до 83%.
Двете диагностични изобразителни техники имат предимства и не-
достатъци. MРТ неврография показва по-добри възможности от ултраз-
вуковото изследване за проследяване еволюцията на промените в диаме-
търа и сигналните промени на нервните коренчета. Не е известно обаче
значението на граничните промени. За провеждането и интерпретация на
резултатите при MРТ неврография се изисква специфична неврорадио-
логична експертиза (48). Ултразвуковото изследване е много по-достъп-
на диагностична методика, може да се извършва при леглото на болния
и е по-флексибилно при възникване на необходимост за включване на
нервите на мишницата. За провеждане и интерпретация на резултатите е
също необходимо специализирано обучение.
Контрастното усилване при MРТ неврография, както и установява-
нето на хиперваскуларизация при ултразвуково изследване не корелират
със специфични възпалителни промени, а са най-вероятно индикатор за
тежестта на хроничния възпалителен процес (39).
3.3. Напреднали биопсични техники
3.3.1. Таргетна фасцикуларна биопсия
Нервната биопсия е важна диагностична процедура при заболяване
на периферните нерви. Повечето невропатии са зависими от дължината
на нервите заболявания, поради което най-изразени промени се наблю-
дават в дисталните сегменти. Най-често се биопсират дистални сензор-
ни нерви, най-често n. suralis. В зависимост от клиничната манифестация
могат да бъдат изследвани и други нерви (n. peroneous superficialis и n.
radialis superficialis). Тези нерви са лесно достъпни, а остатъчният дефи-
цит след процедурата е приемлив и включва основно болка и сензорна
загуба. При невропатии, представящи се основно с моторни симптоми,
се биопсират смесени или чисто моторни нерви, каквито са проксимал-
ният сегмент на n. peroneous superficialis (4), клончета на n. obturatorius за
m. gracilis (3) и пронаторния клон на n. medianus (31).
Диагностичната полза от нервната биопсия е трудно да се прецени,
тъй като се използват различни техники и се оценяват разнообразни па-
тологии. За n. suralis тя варира от 35% до 50% (90, 108). Обяснението за
относително ниската диагностична полза е просто, тъй като не всички
заболявания водят до генерализирана увреда на периферните нерви.
Таргетната биопсия би могла да диагностицира по-локализирани
процеси и може да увеличи диагностичната чувствителност при по-фо-
кално или по-проксимално засягане. През последните десетилетия изо-
бразяването на периферните нерви значително се подобри и се описаха
редица нови абнормности. За първи път Oberlin и съавт. (2009) доклад-
ват биопсична техника, която позволява изследването на отделни фас-
цикули от големи нерви, без това да предизва сигнификантен остатъчен
дефицит (87). В последствие фасцикуларната техника се адаптира за n.
suralis и се комбинира с т.нар. имидж-направлявана биопсия, използвана
преди за диагностика на промени на централната нервна система (105).
Разработиха се специализирани диагностични протоколи, позволяващи
комбинирането на клинични, електрофизиологични, изобразителни и
патохистологични промени. Комбинираният подход дава възможност за
по-добро селектиране на периферно-нервните таргети, носи по-голяма
диагностична полза и безопасност.
Диагностичната полза от таргетната фасцикуларна биопсия на
n.ischiadicus в различни клинични серии е средно 85%, многократно над-
вишаваща конвенционалната биопсия. Това показва, че при подходящо
селектирани пациенти, при които предоперативното изследване насочва
към по-локализирана проксимална патология, ползата от провеждане на
таргентна биопсия е много по-голяма от тази на дистален нерв.
Освен ХВДП (9) таргентната фасцикуларна биопсия на n. ischiadicus
е показа и при: perineuroma (9), lipomatosis (82) и нервно-мускулна
horistoma (86). При съмнение за неоплазия резултатите от таргентна фас-
цикуларна са негативни в около 15% от случаите. Поради тази причина
негативният резултат не винаги изключва инвазия на периферните нер-
ви, особено ако е доказан карцином с друга органна локализация.
Безопастността на тази иначе инвазивна процедура зависи от пре-
оперативната селекция на таргетния нерв, функционалния статус на нер-
ва, броя биопсирани фасцикули, познанията на (интра)невралната анато-
мия, използването на интраоперативен стимулатор.
Целта е провеждане на възможно най-малко рискова процедура по
отношение на постоперативния дефицит и перипроцедурния риск.
Биопсират се възможно най-малък брой нервни фасцикули. Лезиите
на лумбо-сакралния плексус често засягат и интрадуралните нервни ко-
ренчета, както и проксималния сегмент на седалищния нерв. При лум-
бо-сакрална патология биопсирането на n. ischiadicus в областта на седа-
лищния изход е много по-лесен и по-безопасен подход от биопсията на
cauda equina или трансабдоминалната биопсия на плексуса.
Друго важно условие за безопастността на процедурата е резекция
единствено на нефункциониращи или хипофункциониращи нервни струк-
тури. При резекцията на фасцикули, които не отговарят на стимулация,
е възможно да се биопсират както нефункциониращи моторни влакна,
така и сензорни влакна. Когато са патологично променени функциони-
ращи моторни фасцикули и е наложително получаване на спесимени от
тях (напр. при съмнение за малигнизация) се препоръчва резекция на
моторни фасцикули само от тибиалисната порция (вместо от пероне-
алния компонент). Това е така, защото тибиалисният компонент е по-го-
лям, има повече на брой фасцикули, поради което биопсията се последва
по-рядко от значим моторен дефицит.
Когато нозологичните единици се представят с класически невро-
изобразителни белези, не се налага провеждането на таргетна биопсия.
При ХВДП често се наблюдават хиперинтензни в T2 и FLAIR-T2 MRI
секвенции промени в периферните нерви и твърде слабо усилване при
контрастиране с gadolinium.
Таргетната фасцикуларна биопсия не е съвсем безопасна процедура
и понякога се последва от развитие на туморни образувания, каквато е
невромускулната horistoma.
Сравнителните MРТ и биопсични проучвания показват, че невро-
изобразителните промени не винаги корелират с подлежаща патология,
тъй като абнормностите в нервите понякога са резултат от по-прокси-
мална патология (напр. лумбална спондилоза). Ако болестният процес
обхваща повече от един нервен сегмент не се препоръчва провеждането
на биопсично изследване, а се търси по-щадящ подход. Препоръчва се
резекцията на неотговарящи при стимулация фасцикули и резекцията на
максимално дълги фасцикули (поради мултифокалното разпространение
на промените при ХВДП).
Laumonerie и съавт. (2017) са публикували своите резултати при
таргетна фасцикуларна биопсия на брахиалния плексус (72). При нея се
използва супраклавикуларен, инфраклавикуларен или медиален подход
през проксималното рамо. Специфичното място за резекция на фасци-
кули се избира на основа на клиничното, MРТ и електрофизиологично
изследване на брахиалния плексус.
Най-често установяваната патология при този подход е: ХВДП,
neulymphomatosis и perineuroma, метастази от карцином на гърдата и по-
страдиационни промени. При добро познаване на регионалната анатомия
и при използване на стимулационна техника за получаване единствено
на нефункциониращи фасцикули, процедурата не се съпровожда от зна-
чителен дефицит. Тази инвазивна процедура обаче трябва да се извършва
с голямо внимание при екстензивна фиброза на плексуса. Болните с опе-
ративни интервенции и преминали лъчелечение са изложени на висок
риск от компликации като лимфоедема и значим неврологичен дефицит.
3.3.2. Биопсия от кожа: оценка на промените на нодалната
архитектоника
Специфичната молекулярна архитектоника на нодалните региони
осигурява салтаторното провеждане и ключов индикатор за интегритета
на връзките между аксона и Швановите клетки (33). Нодалните участъци
на Ранвие на дермалните нерви за първи път са успешно визуализирани
и количествено анализирани от Doppler и съавт. (2012) (27).
Биопсията от кожа (за разлика от нервната биопсия) е минимално
инвазивна, лесно изпълнима и при необходимост повторима. Кожните
спесимени се получават под локална анестезия със специфичен инстру-
мент (punch биопсия). Инцизира се малко кръгче с диаметър от 5 mm
от кожата на бедрото и 3 mm от обезкосмената повърхност на показале-
ца, между интерфалангелната и палмофалангеална става (74). Кожата на
пръстите позволява анализа на много по-голям брой нодални региони от
тази на бедрото (100). Следва фиксация в 4% формалдехид и маркиране
на замразените срезове с анти-PGP9.5 (аксонален маркер) и анти-MBP
(миелинов маркер), аntipanneurofascin, анти-caspr, антитела срещу Na+
каналчета и вторични флуоресциращи антитела, съгласно описаната от
Saporta и съавт. (2009) и Doppler и съавт. (2012) методика (111).
На основа на имунофлуоресцентна микроскопия е разгадана нодал-
ната архитектоника в кожни спесимени от здрави контроли и са приведе-
ни доказателства за нарушение в нодалните участъци при демиелинизи-
ращи невропатии (27). Спесимените от кожа демонстрират удължаване
на нодалните участъци и нарушение на клъстера на Na+каначнета при
ХВДП и мултиплена склероза (21, 28, 52) и запазена нодална архитекто-
ника при наследствени невропатии (74, 111) и невъзпалителни демиели-
низиращи невропатии (28).
Контактин асоциираният протени (caspr) е полезен диагностичен
маркер за разграничаване на ХВДП от хроничните възпалителни аксо-
нални полиневропатии (21). Спесимените от кожа показват, че диспер-
сията на cspr се наблюдава не е специфична за ХВДП, а се наблюдава
и при невъзпалителни демиелинизиращи невропатии, което показва че
дисперсията cspr е по-скоро маркер за демиелинизация.
Neurofascin-155 e паранодален протеин, изпълняващ ролята на пре-
града между юкстапаранода и нода (115, 120). В нормалните контроли
той е неизменно локализира в инцизурите на Schmidt-Lantermann, пара-
нода и нода (23). Неговата гъстота в периферните нерви е намалена при
експерименталния алергичен неврит (77), а спесимените от кожа на бо-
лни с демиелинизиращи невропатии демонстрират пръсната по хода на
нервите неврофасцинова имунореактивност (27).
4. Нови терапевтични стратегии
В следващите години проучванията при ГБС принципно ще бъдат
насочени в две посоки: оценка на ефективността и безопастността на
инхибиторите на комплемента и подобряване на терапевтичната ефек-
тивност на IVIg.
4.1. Терапевтично приложение на инхибитори на комплемента
Експерименталните проучвания показват значителна редукция на
увредата на периферните нерви след прилагане на инхибитори на ком-
племента: microcept (инхибитор на C3 конвертаза), eculizumab (хумани-
зирано антитяло, залавящо се с голям афинитет към C5 фактора на ком-
племента и блокиращо образуването на цитолитичен C5b-9 комплекс),
nafamostat mesylate (инхибитор на сериновата протеаза и компементната
C1s, C1r, C3a, C3b, C5a, C5b и C5b-9 каскада, който в Япония е използван
за лечение на дисеминирана интраваскуларна коагулация и остър пан-
креатит, в последната година изпитван като антивирусен медикамент, а
в последната година като агент, предотвъртяващ клетъчното проникване
на SARS-CoV-2 (43,44).
Фаза I проучвания на тези терапевтични агенти не се необходими при
GBS, тъй като са провеждани при други индикации (124). Терапевтич-
ната ефективност при GBS може да се изследва в рандомизирани пла-
цебо-контролирани проучвания, чиито протоколи да включват самостоя-
телно или комбинирано (плюс IVIg) приложение на инхибиторите на
комплемента. При установяване на ефективност при GBS индикациите
могат да се разширят и се включат други коплемент-медиирани заболя-
вания: системен еритематоден лупус, дерматомиозит, миастения и опти-
чен невромиелит (124).
4.2. Подобрение на терапевтичната ефективност на IVIg
IgG имуноглобулините са важни за протекция на гостоприемниците
срещу инфекциозни причинители, като в същото време играят важна па-
тогенетична роля за редица автоимунни болести (ревматоиден артрит,
системен лупус, имунотромбоцитопения, автоимунна хемолитична ане-
мия и ХВДП). Същият клас про-инфламаторни молекули се използват и
за противовъзпалително лечение на автоимунните заболявани. В имуно-
логията тези взаимно противоречащи си ефекти на IVIg са известни като
IVIg „парадокс“ (112).
За да се разберат противоположните ефекти на IgG трябва да се по-
знават съвременните разбирания за проинфламаторните механизми.
Изследванията в последните десетилетия потвърждават решаващата
проинфламаторна роля на IgG константния фрагмент (Fc фрагмент),
действащ като своеобразен адаптор за връзка между адаптивнната (нес-
пецифична) и вродената (специфична) имунна система (Фиг. 23). След
свързване на различните IgG молекули към специфичните таргетни ан-
тигени се активират и двете рамена на адаптивната имунна система
(хуморален и клетъчен имунитет) и се отключва проинфламаторен ефек-
торен отговор. Освен това IgG може да инициира вроден имунен отговор
и чрез системата на комплемента, като се активира класическия C1q
зависим път и допълнително към това се активират вродените имунни
клетки чрез свързане към Fc рецепторите (FcyRs). Последният механи-
зъм според in vivo проучвания е доминиращ пред този на комплемента
при повече от автоимунните заболявания.
Съществува цяла фамилия FcyRs, няколко вида активиращи рецеп-
тори и един рецептор с инхибиторна функция (FcyRIIB), експресирани
върху повечето клетки на вродената имунна система – базофили, еози-
нофили, неутрофили, мастни клетки, моноцити и макрофаги. Допълни-
телно към тях, дентритните клетки и B клетките също експресират рe-
цептори от FcyRs фамилия.
Въпреки че повечето FcyRs имат нисък афинитет към мономерни IgG
молекули и могат да бъдат активирани в отговор само на мултимерни IgG
молекули, каквито са патогенните имунните комплекси и димерните фор-
ми на IVIg, някои от рецепторите (FCyR1) имат висок афинитет към IgG
и могат да разпознават определени IgG субкласове в мономерна форма.
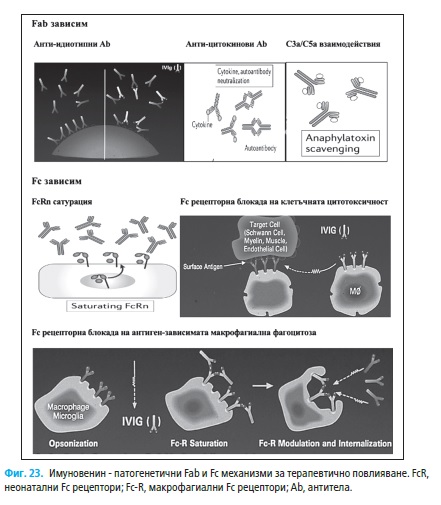
Тъй като IgG имунни комплекси могат да се свързват едноврeменно
към активиращи FcyRs и инхибиращи FcyRIIB, се тригерират едновре-
менно както активиращи, така и инхибиращи клетъчни сигнали, което
играе решаваща роля за нагласяване прага за активация в отговор на IgG.
Експерименталните проучвания показват, че корекцията на FcyRIIB
eкспресия може да предизвика отслабена антитяло-продукция и потиска-
не на автоимунния отговор. Допълнително към това, изолираното свърз-
ване на IgG към FcyRIIB рецептори на B-клетки и плазматични клетки
може индуцира апоптоза, показващо че IgG взема активно участие за
подържане на клетъчната хомеостаза (112).
Редица експериментални проучвания показват, че инфузията на IVIg
води до увеличена експресия на инхибиторни FcyRIIB рецептори върху
мембраните на миелодните клетки, което обяснява антинфламаторното
му действие.
Въпреки значителния напредък в разгадаване на антиинфламаторни-
те механизми, все още остава неясна причината за необходимостта от го-
лемите количества IVIg (1 – 3 g/kg) за постигане на терапевтичен ефект,
а също така и по какъв механизъм IVIg индуцира хиперкспресия на инхи-
биторни FcRIIB рецептори.
В последните години се установи изключителната роля на IgG гли-
козилиране за подържане терапевтичната ефективност на IVIg. Екс-
периментални проучвания показват, че отстраняването от IgG на ас-
паргин-свързана въглехидратна молекула се съпровожда от загуба на
антиинфламаторната активност на IVIg. Серумната гликолизация е хе-
терогенна процес, при който за всеки един от IgG субкласове се гене-
рират повече от 30 различни глюковарианти. Изненадващо се установи,
че третирането на IVIg препарати с ензима неураминидаза, при което
се премахват терминално-свързаните молекули на сиаловата киселина,
намалява IVIg активност, докато обогатяването на IVIg препарати със
сиалова киселина води до 10-кратно по-висока антиинфламаторна актив-
ност. Това най-вероятно се дължи на предизвикани от сиалова киселина
конформационни промени на Fc участъци на IVIg, значително увелича-
ващи афинитета им към Fc рецептори (112).
Доколко тези експериментални открития могат да доведат до подо-
брение на терапевтичната ефективност на IVIg, предстои да се изясни в
бъдещи клинични проучвания.
4.3. Протеозомни и FcRn инхибитори
Основният патогенетичен механизъм при ХВДП включват дисфунк-
ция на имунорегулаторните механизми в периферните лимфоидни ор-
гани, последвана от възпалителна инфилтрация (основно от макрофа-
ги и Т-лимфоцити) на периферните нерви и спинални коренчета (110).
Антиинфламаторният ефект на кортикостероидите при ХВДП се дължи
вероятно на техния директен цитотоксичен ефект върху възпалителните
клетъчни популации и потискане на цитокиновата синтеза (78, 135).
Общите характеристики на използваните при ХВДП имуносупре-
санти включват въздействие върху клетъчния автоимунитет, лимфо-
цитно-активиращите фактори (ciclosporin A), лимфоцитнатата преживяе-
мост (azathioprine и cyclophosphamide), както и блокиране на пуриновата
de novo синтеза на В- и Т клетки (micofenolat mofetil) (32).
Белезите на демиелинизация намаляват след плазмафереза и IVIg,
като експерименталните проучвания демонстрират възможност за па-
сивен трансфер на демиелинизация след инжектиране серум от ХВДП
болни, което недвусмислено потвърждава патогенетична роля на хумо-
ралните фактори (141).
Проучванията на хуморалния имунитет при ХВДП показва, че освен
интернода е възможна и друга локализация на таргетния антиген – но-
далните и паранодални региони. Антителата срещу contactin-1 протеин
предизвикват деструкция на паранода и предизвикват проводен блок,
което предизвиква болезнена възпалителна невропатия. Патологичният
синтез на neurofascin-IgG4 антитела се асоциира с поява на инвалидизи-
ращ тремор и е предиктор за отсъствие на терапевтичен отговор към IVIg
(26, 101). Това налага използване на терапевтични агенти, инхибиращи
генерирането, активацията и функцията на B клетките. Първата линия
медикаменти (IVIg и плазмафереза) не са достатъчно ефективни в тези
случаи, тъй като техния ефект върху антитяловата продукция и функция
е индиректен (79).
В-клетъчно-специфичната инхибиция индуцирана от rituximab
(CD20 таргетно моноклонално антитяло) показва клинично подобрение
в 50% от случаите (вкл. и при наличие на паранодални антитела). Някои
агресивни форми на ХВДП са резистентни на анти-CD20 терапия най-ве-
роятно поради това, че rituximab не повлиява зрелите (с дълъг живот)
плазмоцитни форми, тъй като не експресират CD20 рецептори (66).
Плазматичните клетки преживяват до няколко седмици в периферните
лимфоидни органи и само тези с кратък живот се повлияват от кор-
тикостероидната и цитостатична терапия (122). Експерименталият
модел на Manz и съавт. (1987) показва, че при имунизация с овалбумин
дългоживеещите (>3 месеца) плазмоцити съставляват 70% от плазма-
тични клетки. Въпреки че тези клетки не пролиферират, удълженият им
живот се съпровожда от продължителна секреция на патогенни антитела
(81).
Недостатъците на В клетъчната инхибиция се избягват при приложе-
ние на bortezomib (BTZ), който индуцира апоптоза на зрелите плазма-
тични клетки с дълъг живот.
Терапевтичната ефективност на BTZ е потвърдена при болни с мул-
типлен миелом (99). BZB е селективен и обратим протеозомен инхи-
битор, който при миеломна болест се прилага по специфична химиоте-
рапевтична схема (1,3 mg/m2 s.c. на 1, 4, 8, 11 ден и на всеки 21 ден в 3
или 4 цикъла). Предизвиканата от BTZ ubiquitine-специфична протео-
зомна инхибиция променя хомеостазата на плазматичните клетки като се
потиска разграждането на един ключов протеин, регулиращ клетъчния
цикъл и апоптоза. Протеозомните инхибитори подтискат активирането на
нуклеарния фактор-κB, а също така и секрецията на проинфламаторните
цитокини – тумор некротизиращ алфа фактор и интерлевкин-6. Терапев-
тичният ефект на BTZ e широкоспектърeн и е доказан върху Т-клетки,
В-клетки (плазмоцити), дендритни клетки, обясняващо значителните му
антиинфламаторни възможности (140).
Pitarokoili и съавт. (2017) прилагат BTZ при 10 случая на CIDP, резис-
тентни на ескалираща терапия с rituximab и cyclophosphamide. Приложе-
нието на BTZ е довело до стабилизиране на клиничното състояние при
всички болни, а при част от тях е наблюдавано и подобрение (99).
Специфичните за протеозомните инхибитори неблагоприятни стра-
нични ефекти са сериозна пречка при заболявания на периферните нер-
ви. При болни с мултиплен миелом прилагането BTZ води до развитие
на сензорна аксонална невропатия (96), наблюдаваща се при 30-60% от
лекуваните след 3 или 4 терапевтичен цикъл (85). Експерименталните
проучвания показват, че полиневропатията се дължи на индуциран от
BTZ ендоплазматичен ретикулумен стрес, митотоксичност или увреда
на DNA при наличие на специфична генетична предиспозиция (5).
В проучването на Pitarokoili и съавт. (2017) болните с ХВДП на BTZ
не са показали влошаване на невропатията, вероятно поради по-малкия
брой терапевтични цикли (99). В същия дозов режим BTZ е използван
и при автоимунни заболявания на централната нервна система, където
също не e регистрирана невропатия. Въпреки това потенциални странич-
ни ефекти сериозно лимитира дозовия режим на BTZ при резистентни
ХВДП случаи.
Очаква се скоро да излязат резултатите на фаза I и II проучванията
на rozanolixizumab (IgG4 моноклоналното антитяло) и efgartigimod
(IgG1 Fc фрагмента) при болни с ХВДП. Такива проучвания се про-
веждат и при миастения, където са в по-напреднала II и III фаза. И два-
та терапевтични агента имат сходен механизъм на дейстие, а именно
блокиране на неонаталните FcRn рецептори (112). Тези рецептори в
здравите индивиди подържат хомеостазата на IgG (Фиг. 24A), като трас-
портират непроменени молекулите на IgG през клетъчните мембрани и
предотвъртяват ендозомалната деструкция. При блокиране на FcRn (с
имуновенин или моноклонални антитела), свободните патогенни анти-
тела се подлагат на вътреклетъчна деструкция (Фиг. 24B). Доколко този
имунологичен механизъм би могъл да бъде използван при ХВДП и миас-
тения за продуциране на терапевтичен ефект („вътреклетъчна плазмафе-
реза“) ще стане ясно в следващите години.